Introduction
Cholestasis, one of the most common presentations of
liver disease, is the major cause of severe liver dysfunction in
childhood, which eventually necessitates liver transplantation
(1). Despite extensive
investigation, the etiology and pathogenesis of cholestasis in a
significant proportion of children remains unknown. Recent advances
in molecular biology have elucidated the genetic basis for a
subgroup of disorders, including progressive familial intrahepatic
cholestasis (PFIC). PFIC is a heterogeneous group of autosomal
recessive disorders characterized by early onset cholestasis,
progressive liver cirrhosis and hepatic failure in early childhood
(2). According to the genetic
defects, three types of PFIC have been identified. PFIC1, PFIC2 and
PFIC3 are caused by mutations in ATP8B1 (encoding FIC1), ABCB11
[encoding bile salt export pump, (BSEP)] and ABCB4 [encoding
multidrug resistant protein 3, (MDR3)] genes, respectively
(3). Although the actual
prevalence of PFIC remains unknown, the estimated incidence is
1/50,000–100,000 births worldwide (4).
The ABCB11 gene located on chromosome 2q24 encodes
an ATP-binding cassette (ABC) transporter, known as BSEP in human
liver. BSEP is selectively expressed in the hepatocyte canalicular
membrane and is the major exporter of bile salts against extreme
concentration gradients, in an ATP-dependent manner (5). Mutations in this gene are responsible
for decreased biliary bile salt secretion, which leads to decreased
bile flow and the accumulation of bile salts inside the
hepatocytes, thereby resulting in a spectrum of cholestatic disease
(6). This ranges from the severe
phenotype of PFIC2 (7) to milder,
intermittent forms of cholestasis, including benign recurrent
intrahepatic cholestasis type 2 (8), drug-induced cholestasis (DIC)
(9), intrahepatic cholestasis
pregnancy (ICP) (10) and
contraceptive-induced cholestasis (11). Furthermore, two significant single
nucleotide polymorphisms (SNPs) of ABCB11 have been associated with
non-cholestatic and cholestatic liver disease. One is V444A (HGVS
name: NM_003742.2:c.1331T>C; refSNP: rs2287622) in exon 13,
which has been reported to be correlated with cholestasis (12) and chronic hepatititis C virus
infection (13). Furthermore, the
polymorphism was able to reduce the levels of mature BSEP (14). The other is A1028A (HGVS name:
NM_003742.2:c.3084A>G; refSNP: rs497692) in exon 24. It has been
described to cause altered splicing events of ABCB11 through severe
exon skipping in vitro (14) and possibly associates with primary
biliary cirrhosis (PBC) (15).
Whether they are associated with cholestatic liver disease;
however, remains controversial (11,16).
Clinical and biochemical characteristics of patients
with ABCB11 mutations in early infancy, include jaundice, pruritus,
growth failure, hepatomegaly, splenomegaly, complications due to
fat-soluble vitamin deficiency, increased serum bilirubin
concentrations particularly direct bilirubin, normal or low serum
γ-glutamyltransferase (GGT) and high serum bile acid concentrations
(6). These symptoms are not
specific and often overlap with those of other cholestatic liver
diseases, such as biliary atresia, inherited cholestatic disease,
neonatal hepatitis and metabolic diseases, rendering prompt and
accurate clinical diagnosis particularly difficult. Previously,
mutations in ABCB11 have been detected predominantly in European
and American patients. Studies involving Asian patients,
particularly Chinese subjects, are notably lacking (16,17).
Current diagnostic methods used for inherited intrahepatic
cholestasis include clinical features, biochemical parameters,
liver histology and genetic analysis. These lack proficient
accuracy, are time consuming and expensive or are not widely
available. High-resolution melting (HRM) analysis prior to
sequencing has been described as an effective, sensitive and
economical method for detecting genetic variations (18).
The present study aimed to apply HRM analysis prior
to sequencing to identify genetic variations more efficiently in
the ABCB11 gene in Chinese patients. This screening approach allows
a faster and more economical diagnosis to be made in patients
suspected of carrying ABCB11 mutations.
Materials and methods
Patients and DNA samples
All of the subjects were recruited prospectively at
the Nanjing Children’s Hospital Affiliated to Nanjing Medical
University (Nanjing, China) between July 2010 and June 2013. All
patients were <1 year old and suffered from cholestasis,
presenting with elevated conjugated hyperbilirubinaemia or
pruritus. Basic demographic data of the patients and blood
biochemistry examinations were reviewed from the medical records. A
total of 20 patients (numbered P1–P20) were included for detection
of ABCB11 mutations. The inclusion criteria were as follows: (i)
Onset of conjugated hyperbilirubinaemia or pruritus prior to 12
months of age. Conjugated hyperbilirubinaemia was defined as a
direct bilirubin level of 20% of the total bilirubin or >17.1
μmol/l if the total serum bilirubin was <85.5 μmol/l (17); (ii) the serum GGT levels were
consistently below the normal value for infancy (<94 u/l)
(19); (iii) clinical
manifestations, including jaundice, pruritus, growth failure,
hepatomegaly or splenomegaly were observed and (iv) a thorough
search for the known causes of cholestasis in infancy had been
negative, including viral or bacterial infection and inborn errors
of bile acid synthesis. Patients with biliary tree anomalies were
excluded.
All of the DNA samples were extracted from
peripheral EDTA-anticoagulated whole blood of all the patients and
some parents using a TIANamp Blood DNA kit (Tiangen Biotech
(Beijing) Co., Ltd, Beijing, China), according to the
manufacturer’s instructions. Informed consent was obtained from all
of the parents of the infants involved in the study. The study was
approved by the Institutional Review Board of Nanjing Children’s
Hospital Affiliated to Nanjing Medical University (Nanjing,
China).
Polymerase chain reaction (PCR)
amplification of exons of ABCB11
A total of 27 sets of primers were designed to
obtain amplicons with a size ≤400 bp (range, 113–321) for the
coding sequences and splice sites (ten nucleotides away from the
exon) of exon 2–28 of the ABCB11 gene (Table I). The concentrations of DNA
samples were measured using the NanoDrop 2000 UV-Vis
Spectrophotometer (Thermo Scientific, Wilmington, DE, USA), then
the DNA samples were diluted to 10 ng/μl. PCR was performed in a
total volume of 10 μl containing 30 ng genomic DNA, 3 pmol of each
primer, 1 μl of 10X Taq Buffer with
(NH4)2SO4, 1 μl of dNTP Mixture
(2.5 mM each), 1 μl of 25 mM MgCl2, 0.4 units Taq
DNA Polymerase (recombinant; Thermo Scientific), 1 μl of 1X
LCGreen® PLUS (Idaho Technology, Salt Lake City, UT,
USA) and water (molecular grade) added up to 10 μl. A PTC-200 DNA
Engine® Peltier Thermo Cycler (Bio-Rad, Hercules, CA,
USA) was used for PCR with the following cycling conditions: An
initial denaturation step at 95°C for 5 min, followed by 40 cycles
consisting of denaturation at 95°C for 30 sec, annealing at 61°C
for 30 sec, extension at 72°C for 20 sec and a final extension step
at 72°C for 10 min followed by cooling to 25°C for 5 min.
 | Table IPrimers for PCR amplification of
ABCB11 for HRM analysis. |
Table I
Primers for PCR amplification of
ABCB11 for HRM analysis.
| Exon | Primer
sequence | Amplicon length
(bp) |
|---|
| 2 | F: 5′
CTTTCGTTTGGCTACTTTGATTA 3′
R: 5′ TGTACAAGATGCAGTGAGG 3′ | 213 |
| 3 | F: 5′
CGTTGCATTTTGTCATTATTATTAACT 3′
R: 5′ AAAGATGCTGCATTGTTGAA 3′ | 113 |
| 4 | F: 5′
ATTGTATTGGAAAGGGTGGTC 3′
R: 5′ TATAGCTGCACACCCACT 3′ | 144 |
| 5 | F: 5′
AGATGATCTCTGAACCCTT 3′
R: 5′ TTGAATTACAATTTAAGATATGAGCAA 3′ | 329 |
| 6 | F: 5′
CAAGTCTGAACATTCTTTTCCC 3′
R: 5′ CACATTGCATCTCATTGTAGTG 3′ | 191 |
| 7 | F: 5′
TGCAATGCTAAACATTCCTT3′
R: 5′ AAGGGTTTTATTATCCAAAAATCAC3′ | 223 |
| 8 | F: 5′
ACTGAGACTTTCAGCAAGATA 3′
R: 5′ CCCATGGAGAGATGCAA 3′ | 274 |
| 9 | F: 5′
CTTACCTAATTTCTTGGACTTCACA 3′
R: 5′ ACTATGCTGATTGATGAAATTAAGGA 3′ | 228 |
| 10 | F: 5′
TAACTTGAGCTGTTTCTGCC 3′
R: 5′ ATGTCTCGGTCAATAAGTCC 3′ | 252 |
| 11 | F: 5′
CATGGAAGACCCAAATGATAGT 3′
R: 5′ CCCCACCTGTTAATGGC 3′ | 233 |
| 12 | F: 5′
TAGTTTGAGTTTACACTGTGTCC 3′
R: 5′ ATCATCGAAGAAGAAAACATTTACTAT 3′ | 204 |
| 13 | F: 5′
GTGACAATCTGAACTTTGCT 3′
R: 5′ TGATCAATATACTGCCATTTGC 3′ | 231 |
| 14 | F: 5′
GTTGTGATGTTGTGCCC 3′
R: 5′ TTCCTTCTATGACCTCTTAGTTT 3′ | 282 |
| 15 | F: 5′
CAGAAGCCATCAAATTCTTT 3′
R: 5′ GCATTTCCACATGGACC 3′ | 268 |
| 16 | F: 5′
TTTGTTTAATGGTGCACTGT 3′
R: 5′ AAACCGTAAAGCACTATAGAC 3′ | 301 |
| 17 | F: 5′
CTTGGATATGGTTCTGTTTATTGTA 3′
R: 5′ AAAGCTTGTAATCTGCCC 3′ | 157 |
| 18 | F: 5′
GTCTACACTGTTTCATCTTTCTG 3′
R: 5′ CAGGAGAGACTTCTTCCATTC 3′ | 184 |
| 19 | F: 5′
ATGTCTTGAGTACATTTAGATGAT 3′
R: 5′ TGAGAAGAAGAAAGCTAGTCCA 3′ | 249 |
| 20 | F: 5′
TTGGACAGATATATAATGACATGGT 3′
R: 5′ AAGGAAAAATAACTAAATCACTTACTG 3′ | 258 |
| 21 | F: 5′
AATTTCTCTAACATCTCCCTCT 3′
R: 5′ AGAATGCCAATGCAGTTAATATAC 3′ | 244 |
| 22 | F: 5′
GTGTCTGAGACGGGTTGATT 3′
R: 5′ TCCTTCAGTCTCTTCGTACT 3′ | 331 |
| 23 | F: 5′
TGCCCTTGTATTCCTAAGACTC 3′
R: 5′ ATTCCTTCCTTGTGTGTTGAT 3′ | 330 |
| 24 | F: 5′
GTCTGGTTACAGGGTGATCT 3′
R: 5′ GCTGCATAGTATTCCAACAC 3′ | 193 |
| 25 | F: 5′
GCTTCAGTAAGAGCATCTCTAAT 3′
R: 5′ TTTAGGGGTTGGAAATACTCTG 3′ | 300 |
| 26 | F: 5′
AAACCTAATGACCTGTCATCTC 3′
R: 5′ ATAGGGAATGGCTCTGACT 3′ | 270 |
| 27 | F: 5′
AGGAGCAATAACTGTTTCTATTT 3′
R: 5′ AGACTTATTTGTAATGATCTAAGACT 3′ | 241 |
| 28 | F: 5′
GCATCTTTGCATCAACTTTC 3′
R: 5′ TAACTGGTGCGTCATGT 3′ | 277 |
HRM analysis
Following completion of PCR, 10 μl of the PCR
products were added into a 96-well plate (Bio-Rad). The mixtures
were overlaid with 20 μl of mineral oil (Sigma-Aldrich, St. Louis,
MO, USA) and the plate was centrifuged at 2,000 × g for 1 min. The
plate was transferred to the LightScanner® (Idaho
Technology), with fluorescence data collection over the temperature
range 70–97°C, as samples were melted.
In all instances, HRM directly discriminates the
heterozygotes and homozygotes, but not the major and minor
homozygotes of a polymorphism. Next, genotyping using spike-in
control DNA was performed to allow distinction of minor homozygotes
from major homozygotes. Briefly, the PCR products that were
demonstrated to be homozygous were mixed with an equal quantity of
DNA products from a known major allele homozygous subject to apply
to the LightScanner. To promote heteroduplex formation, the
mixtures were firstly centrifuged at 2,000 × g for 1 min, then
denatured in a PTC-100® Peltier Thermo Cycler (Bio-Rad)
under the following conditions: 95°C for 2 min, 25°C for 1 min,
95°C for 2 min, 25°C for 2 min. This strategy converted the minor
allele homozygous form into the heterozygous form, but did not
change the major allele homozygous samples, providing the
distinction between the major allele homozygous samples and the
minor ones. Each sample was analyzed at least three times and all
of the samples demonstrated reproducible results.
Direct sequencing
The samples with shifted melting curves, as
identified by HRM, were amplified through another PCR reaction with
different primer pairs to achieve ~500-bp PCR products for direct
sequencing (Table II). The
reaction mixture in a total of 50 μl contained 50 ng of genomic
DNA, 20 pmol of each primer, 5 μl of 10X Ex Taq Buffer
(Mg2+), 4 μl dNTP mixture (2.5 mM each), 1 unit of Ex
Taq DNA Polymerase (Hot Start; Takara Bio Inc., Dalian,
China) and water (molecular grade) added up to 50 μl. The PCR
reaction was performed under the following conditions: Initial
denaturation at 95°C for 5 min; 40 cycles of 94°C for 30 sec, 59°C
for 30 sec, 72°C for 40 sec and one cycle of 72°C for 10 min. The
PCR products were separated on 2% agarose gels, purified with a gel
extraction kit (Tiangen Biotech (Beijing) Co., Ltd). Direct
sequencing of the purified products was performed in a 3130 Genetic
analyzer (Applied Biosystems, Foster City, CA, USA) using an ABI
BigDye® Terminator v3.1 Cycle Sequencing kit (Applied
Biosystems). All of the tests were performed in duplicate. The
sequences obtained were compared with the ABCB11 reference sequence
(NM_003742.2) derived from publicly available databases provided by
NCBI (http://www.ncbi.nlm.nih.gov/).
| Table IIPrimers for PCR amplification of
ABCB11 for direct sequencing. |
Table II
Primers for PCR amplification of
ABCB11 for direct sequencing.
| Exon | Primer
sequence | Amplicon length
(bp) |
|---|
| 2 | F: 5′
GACTGTGGCTTATCTTTCCTG 3′
R: 5′ CGTTACATGGATTCTAGGGAG 3′ | 461 |
| 3 | F: 5′
GAGTAAAGGTAGCAGCACTC 3′
R: 5′ GGGGACATTTGAACCTAACC 3′ | 500 |
| 4 | F: 5′
CGCTAGTGAACCTGAGATTG 3′
R: 5′ GATAACCATGGGCTTAGTGA 3′ | 519 |
| 5 | F: 5′
CTCTGCCACTCAATTAAGGTG 3′
R: 5′ GAAGGAAACTTGAGGCAGAG 3′ | 550 |
| 6 | F: 5′
GGTACCATGAGGTCTGTTTAG 3′
R: 5′ CAGACTGTAGTTCTTAGGGC 3′ | 431 |
| 7 | F: 5′
CCTGCTGAAGGTTCTGTTTA 3′
R: 5′ ACACACCAAATTGCAGTACC 3′ | 543 |
| 8 | F: 5′
GATCTGAGAGGCTGTTAATGC 3′
R: 5′ GTTGCTAACTGTACTCAGGA 3′ | 414 |
| 9 | F: 5′
CCCTGGATGAAGCTTACCAT 3′
R: 5′ CATACTGCTAAAGGCTTGGG 3′ | 504 |
| 10 | F: 5′
AGTATCGCCCTTTCAACATG 3′
R: 5′ GATGCTTTTTTCCTGAAGGC 3′ | 494 |
| 11 | F: 5′
CCAAACAGCCAAAGAGCTAG 3′
R: 5′ AGTGTTGCTGAATTAAGGGC 3′ | 392 |
| 12 | F: 5′
CAGAGCAACAACCAGATAAAAC 3′
R: 5′ CAACACCCGAGGATACTTTC 3′ | 387 |
| 13 | F: 5′
TACTTCTTGGTCATGGCTCT 3′
R: 5′ GTTACCATGTAGGAAGCGTG 3′ | 532 |
| 14 | F: 5′
GCCTCTATTTTTTCTGCCCAT 3′
R: 5′ GATGAAAGGAAACACTCATGG 3′ | 467 |
| 15 | F: 5′
GTCTGGGGAAGGGATATTTC 3′
R: 5′ TGAGGAAGATTGTAGTCAGC 3′ | 491 |
| 16 | F: 5′
TGTGCTGGCCTTTTCTAATG 3′
R: 5′ CAGAGTTGTTGGGAGAACAG 3′ | 461 |
| 17 | F: 5′
TAGAATCTGCAGGACAAGTC 3′
R: 5′ TCCCCAAGAAGATGAGAAGC 3′ | 451 |
| 18 | F: 5′
CACTCTGAATCTGGGTCCAA 3′
R: 5′ GTCTGACTTGAAACACTGCT 3′ | 463 |
| 19 | F: 5′
ATTCAAGCCACAGCAATAGT 3′
R: 5′ CTTCTTACCCTCTGTGTGATG 3′ | 522 |
| 20–21 | F: 5′
CACAGATCCACAGCTTACAT 3′
R: 5′ ACTGGTCCCTATTCCATAGA 3′ | 624 |
| 22 | F: 5′
ACATTGTGAAATGCCACTGA 3′
R: 5′ AGCTTCCTTCAGTCTCTTCG 3′ | 465 |
| 23 | F: 5′
CTTTGTATTCCCAGATGATGC 3′
R: 5′ TGATGACCCACAGAATCTTG 3′ | 537 |
| 24 | F: 5′
CTCTCCATTTCCAGACAAGT 3′
R: 5′ CTGTGTCCATGTGTTCTGTT 3′ | 483 |
| 25 | F: 5′
CAGAACACAAAATGGAATGTCC 3′
R: 5′ TAGAATCAGGTGAAGCAGCA 3′ | 539 |
| 26 | F: 5′
GCCTTGGGATTGTTAGTCTG 3′
R: 5′ CTGTGGAATCATGTTGGCAT 3′ | 486 |
| 27 | F: 5′
TGCTTCCCACATCAAATGTC 3′
R: 5′ GGTTCCACAAAGTATTGCCA 3′ | 490 |
| 28 | F: 5′
CAGGTCGTGTTAACTGAACT 3′
R: 5′ GCTTGGATTCCGATGTAGGA 3′ | 448 |
To avoid false-negative results from HRM screening,
confirmative direct sequencing was performed for exons 13 and 24 in
which SNPs V444A (in exon 13) and A1028A (in exon 24) were common
and rendered the interpretation of melting patterns difficult. For
the remaining 25 exons, 250 amplicons (50%) without melting curve
shifting were randomly selected for direct sequencing. In total,
the results of 290/540 amplicons from HRM analysis were confirmed
by direct sequencing.
Prediction of functional
consequences
The variants confirmed by direct sequencing were
firstly submitted to the MutationTaster program to evaluate the
disease-causing potential of sequence alterations (http://www.mutationtaster.org/) (20). To predict the functional
consequences of the missense mutations and polymorphisms
identified, three bioinformatics tools based on different
computational methods were applied. Sorting Intolerant From
Tolerant (SIFT) is a program that predicts whether an amino acid
substitution affects protein function, through analysis of an
alignment of orthologous sequences (http://sift.jcvi.org/) (21). A SIFT score of <0.05 indicates
the presence of evolutionarily conserved amino acids, and mutations
in these residues are predicted to be deleterious (22). Polymorphism Phenotyping version 2
(PolyPhen-2) uses empirically derived rules to predict the possible
impact of an amino acid substitution on the structure and function
of a human protein (http://genetics.bwh.harvard.edu/pph2/) (23). It calculates a position-specific
independent count (PSIC) profile for each candidate mutation and
qualitatively predicts whether it is benign, possibly damaging or
highly likely damaging, according to the posterior probability
intervals (0, 0.2), (0.2, 0.85) and (0.85, 1), respectively
(22). SNPs&GO characterized
by statistical accuracy is a web server for the prediction of human
disease-related single point protein mutations, by collecting a
unique framework of information derived from protein sequences,
protein sequence profiles and protein function (http://snps-and-go.biocomp.unibo.it/snps-and-go/)
(24). The mutations effecting
amino acid sequence were estimated to be neutral or
disease-related. The amino acid changes were also classified as
evolutionarily conserved (EC) or non-conserved (EN), based on
sequence alignments with six mammalian orthologs (bonobo, mouse,
rat, rabbit, dog, cattle). It has been demonstrated that
substitutions at evolutionarily conserved positions are more
deleterious than those at evolutionarily non-conserved positions
(25). In order to understand the
prediction of functional consequences, the schematic view of BESP
with the location of the variants identified in the present study
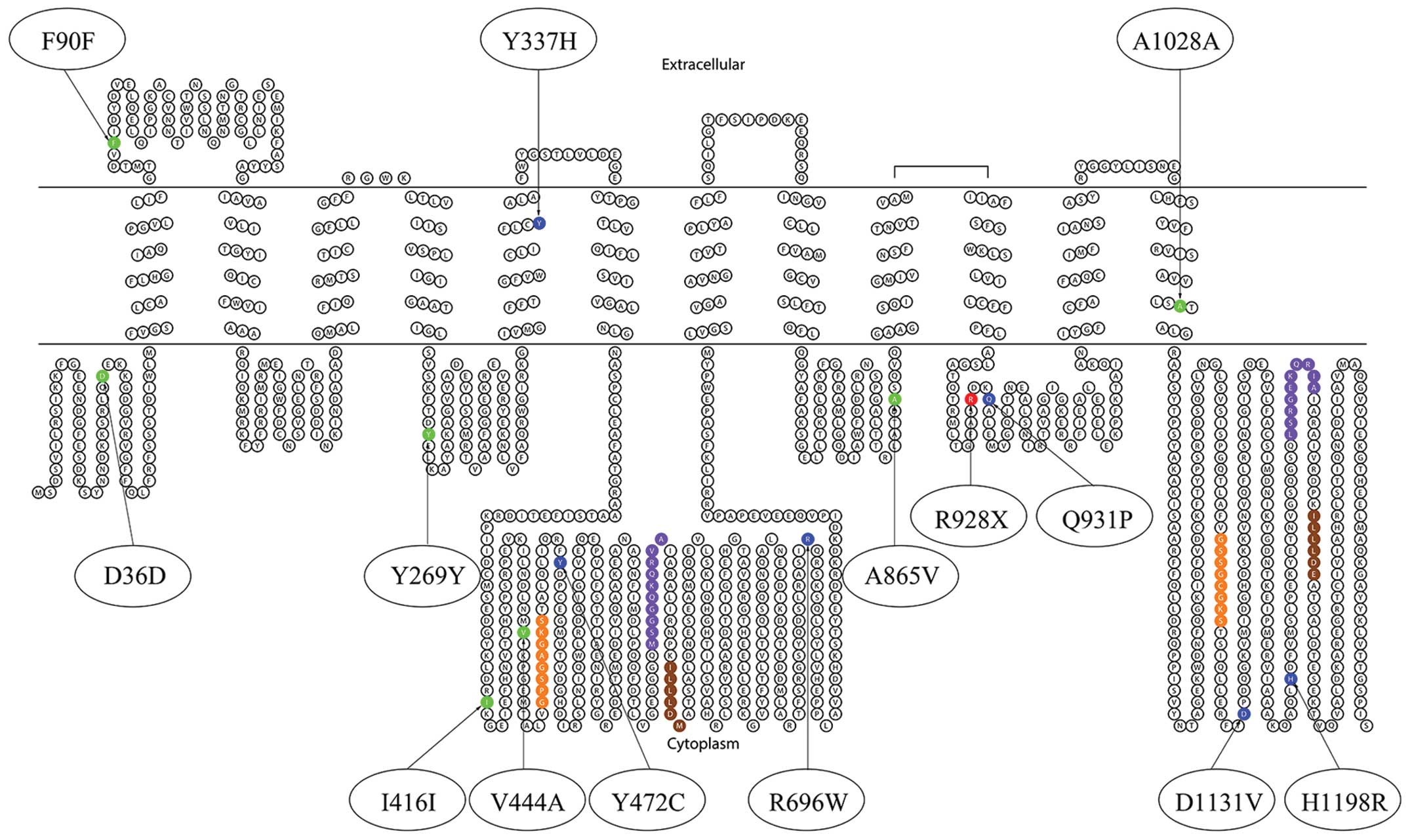
was produced using the TOPO program (http://www.sacs.ucsf.edu/TOPO2/).
Analysis of mutation carrier rate in
control subjects
To determine the carrier rate of mutations
identified, the exons in which mutations existed were screened
using HRM analysis of 200 control subjects from the visitors to the
Nanjing Children’s Hospital Affiliated to Nanjing Medical
University (Nanjing, China). All of the controls were ≤1 year old
and had normal liver and biliary function. All of the exons with
abnormal melting patterns and 50% of the exons with normal melting
patterns were confirmed by direct sequencing.
Analysis of V444A and A1028A in patients
and controls
To investigate whether the two common polymorphisms
found in the study population have functional consequences and
affect disease presentation, the allele frequencies of
polymorphisms were analyzed in 20 patients and in the 200 control
subjects mentioned above. These samples were tested using HRM
analysis of exons 13 and 24, followed by direct sequencing to
identify the V444A and A1028A polymorphisms. Statistical analyses
were performed using Stata 10 (Stata Corporation, College Station,
TX, USA). Associations of categorical variables were tested by
Pearson’s χ2 test. Allele frequencies were tested to
determine whether they were in Hardy-Weinberg equilibrium. All
tests were two-sided and P<0.017 was considered to indicate a
statistically significant difference according to the Bonferroni
correction for multiple testing.
Results
Mutations and SNPs detected in
patients
Among the 20 patients with cholestasis, 14 types of
variants were detected, including seven mutations in the coding
region (p.Y337H, p.Y472C, p.R696W, p.R928X, p.Q931P, p.D1131V and
p.H1198R) and seven SNPs (p.D36D/rs3815675, p.F90F/rs4148777,
p.Y269Y/rs2287616, p.I416I/rs183390670, p.V444A/rs2287622,
p.A865V/rs118109635 and p.A1028A/rs497692). Missense mutations
p.Y337H, p.R696W, p.Q931P, p.D1131V and p.H1198R were novel
mutations identified in the study. All of the mutations and SNPs
identified in the study are summarized in Table III and Fig. 1. The shifted melting curves of
mutated samples are presented in Fig.
2.
| Table IIIMutations and SNPs of the ABCB11 gene
in patients. |
Table III
Mutations and SNPs of the ABCB11 gene
in patients.
| Variant | Exon | Amino acid
change | RefSNP | Patients and
status | Carrier rate in
control (%) |
|---|
| c.108T>C | 4 | D36D | rs3815675 | Heterozygous: P7,
P11, P16 | - |
| c.270T>C | 5 | F90F | rs4148777 | Heterozygous: P6,
P13 | - |
| c.807T>C | 9 | Y269Y | rs2287616 | Heterozygous: P7,
P11, P16 | - |
| c.1009T>C | 10 | Y337H | - | Heterozygous:
P5 | 0 |
| c.1248C>A | 12 | I416I | rs183390670 | Heterozygous:
P13 | - |
| c.1331T>C | 13 | V444A | rs2287622 | Heterozygous: P1,
P5, P12, P16, P17, P19
Homozygous: P2, P3, P4, P6, P7, P8, P9, P10, P11, P14, P15, P18,
P20 | 94.5 |
| c.1415A>G | 13 | Y472C | - | Heterozygous:
P3 | 0 |
| c.2086C>T | 18 | R696W | - | Heterozygous:
P11 | 0 |
| c.2594C>T | 21 | A865V | rs118109635 | Heterozygous: P7,
P17 | - |
| c.2782C>T | 22 | R928X | - | Heterozygous:
P1 | 0 |
| c.2792A>C | 22 | Q931P | - | Heterozygous:
P4 | 0 |
| c.3084A>G | 24 | A1028A | rs497692 | Heterozygous: P1,
P8, P12, P13, P15, P16, P17, P20
Homozygous: P2, P3, P4, P5, P6, P7, P9, P10, P14, P18, P19 | 90.5 |
| c.3392A>T | 25 | D1131V | - | Heterozygous:
P3 | 0 |
| c.3593A>G | 26 | H1198R | - | Heterozygous:
P1 | 0 |
The results based on comprehensive evaluation of
SIFT, PolyPhen-2, SNPs&GO and evolution conservation indicated
that p.Y337H, p.Y472C, p.R696W, p.D1131V and p.H1198R were likely
damaging, p.Q931P and p.A865V were possibly damaging and p.V444A
was predicted to be benign. With the nonsense mutation, p.R928X is
able to introduce a premature stop codon that results in premature
protein truncation or failure of protein production, which had been
reported previously in PFIC2 patients (26). Four patients (P1, P3, P5 and P11)
were identified to have PFIC2 based on the comprehensive
consideration of clinical features and genetic analysis. The
consequences of function prediction of missense mutations and
polymorphisms for BSEP are demonstrated in Table IV.
| Table IVPrediction of functional consequences
of missense mutations and polymorphisms in ABCB11 gene found in
patients. |
Table IV
Prediction of functional consequences
of missense mutations and polymorphisms in ABCB11 gene found in
patients.
| Variant | SIFT | PolyPhen-2 | SNPs&GO | EC/EN |
|---|
| c.1009T>C
(Y337H) | 0.01 | 0.996 | Disease | EC |
| c.1331T>C
(V444A) | 0.34 | 0.001 | Neutral | EC |
| c.1415A>G
(Y472C) | 0 | 1.000 | Disease | EC |
| c.2086C>T
(R696W) | 0.02 | 0.999 | Disease | EC |
| c.2594C>T
(A865V) | 0.07 | 0.880 | Disease | EC |
| c.2792A>C
(Q931P) | 0.02 | 0.178 | Disease | EN |
| c.3392A>T
(D1131V) | 0 | 1.000 | Disease | EC |
| c.3593A>G
(H1198R) | 0 | 1.000 | Disease | EC |
Among the four patients with disease-causing
mutations (P1, P3, P5 and P11), two were heterozygous and two
exhibited compound heterozygous mutations (Table V). Except for P5 and P11 whose
parents’ samples were not available, the parents of P1 and P3
carried one allele of the respective mutation each. The two SNPs of
p.V444A and p.A1028A were also detected in the four patients,
except that P11 only had p.V444A. The detection rate of
disease-causing mutations in ABCB11 was 4/20 (20%) patients from
different families.
| Table VCharacteristics of patients with
disease-causing mutations in ABCB11. |
Table V
Characteristics of patients with
disease-causing mutations in ABCB11.
| Patient | Age of
onset/gender | Symptoms | GGT (U/l) | TBA (μmol/l) | TBIL/DBIL
(μmol/l) | ALT/AST (U/l) | Mutation | Mutation
origin |
|---|
| P1 | 1 m/M | Persistent
jaundice, hepatosplenomegaly | 49 | 101.3 | 162.5/130.4 | 432/606 | Compound
heterozygous p.R928X/p.H1198R | R928X, maternal;
H1198R, paternal. |
| P3 | 2 d/F | Persistent
jaundice, pruritus, hepatosplenomegaly | 32 | 256.1 | 166.7/137.4 | 158/235 | Compound
heterozygous p.Y472C/p.D1131V | Y472C, paternal.
D1131V, maternal. |
| P5 | 6 d/M | Progressive
jaundice | 46 | NA | 99.7/72.6 | 165/211 | Heterozygous
p.Y337H | NA |
| P11 | 4 d/M | Progressive
jaundice | 74 | 204.6 | 75.4/54.3 | 481/600 | Heterozygous
p.R696W | NA |
Rates of false-positive calls and
false-negative calls by HRM analysis
Among the 540 amplicons from the 20 patients, 58
amplicons demonstrated melting curve shifting and 56 (96.6%) of
these samples were found to have mutations by direct sequencing.
The false-positive call rate was 0.37% (2/540) by HRM analysis. As
for the rates of false-negative calls, no mutations or
polymorphisms detected from direct sequencing were missed by the
HRM analysis of the 290 amplicons consisting of the two exons with
common SNPs in the Chinese population (exons 13 and 24) and the
randomly selected 250 amplicons from the remaining 25 exons that
did not demonstrate melting curve shifting. The false-negative rate
was zero.
Mutation carrier rate in control
subjects
All the disease-related mutations detected above
were tested in 200 control subjects using HRM analysis to screen
exon 10 (p.Y337H), exon 13 (p.Y472C), exon 18 (p.R696W), exon 22
(p.R928X), exon 22 (p.Q931P), exon 25 (p.D1131V) and exon 26
(p.H1198R). No distinct mutation patterns were noted in all of the
200 control subjects.
Polymorphism analysis
Two previously reported SNPs, p.V444A and p.A1028A,
were identified in a number of the patients and control subjects.
These two polymorphic sites were examined with HRM analysis
followed by direct sequencing in four patients with PFIC2, 16
patients with cholestasis of undefined etiology from the 20
patients who were not PFIC2, and 200 control patients. The
distribution of p.V444A and p.A1028A polymorphisms, as well as
allele frequencies, are revealed in Table VI. It was identified that V444A
and A1028A were more prevalent polymorphisms in the control
subjects, and had an allele frequency of 74.5 and 67.2%,
respectively. This was consistent with previously reported data,
which identified an allele frequency of 75.6% for V444A (19). However, neither V444A nor A1028A
were associated with PFIC2 or cholestasis of undefined etiology in
the patients. The distribution of alleles at the two SNPs in
control group was in Hardy-Weinberg equilibrium (V444A, P=0.46;
A1028A, P=0.43).
| Table VIDistribution of polymorphisms and
allele frequencies in Chinese patients and control subjects. |
Table VI
Distribution of polymorphisms and
allele frequencies in Chinese patients and control subjects.
| A, p.V444A
(c.1331T>C) |
|---|
|
|---|
| Variable | PFIC2 (%) | Cholestasis
(non-PFIC2) (%) | Control (%) | Pa | Pb | Pc |
|---|
| Polymorphism | | | | 0.847 | 0.493 | 0.580 |
| TT | 0 (0.0) | 1 (6.3) | 11 (5.5) | | | |
| TC | 2 (50.0) | 4 (25.0) | 80 (40.0) | | | |
| CC | 2 (50.0) | 11 (68.7) | 109 (54.5) | | | |
| Total no. of
patients | 4 | 16 | 200 | | | |
| Allele
frequency | | | | 0.974 | 0.396 | 0.693 |
| T (%) | 2 (25.0) | 6 (18.8) | 102 (25.5) | | | |
| C (%) | 6 (75.0) | 26 (81.2) | 298 (74.5) | | | |
|
| B, p.A1028A
(c.3084A>G) |
|
| Variable | PFIC2 (%) | Cholestasis
(non-PFIC2) (%) | Control (%) | Pa | Pb | Pc |
|
| Polymorphism | | | | 0.500 | 0.361 | 0.116 |
| AA | 1 (25.0) | 0 (0.0) | 19 (9.5) | | | |
| AG | 1 (25.0) | 7 (43.8) | 93 (46.5) | | | |
| GG | 2 (50.0) | 9 (56.2) | 88 (44.0) | | | |
| Total no. of
patients | 4 | 16 | 200 | | | |
| Allele
frequency | | | | 0.777 | 0.204 | 0.361 |
| A (%) | 3 (37.5) | 7 (21.9) | 131 (32.8) | | | |
| G (%) | 5 (62.5) | 25 (78.1) | 269 (67.2) | | | |
Discussion
Numerous PCR-based mutation detection methods have
been used to screen ABCB11 mutations. Methodologies described in
the literature include the use of single-strand conformation
polymorphisms (27), denaturing
high performance liquid chromatography (19), gene chips (28), TaqMan probes (29), microsatellite markers (27), restriction fragment length
polymorphisms (27) and DNA
sequencing (17). All of these
methods have their advantages and disadvantages in terms of
simplicity, sensitivity, specificity, time required and
expense.
Direct sequencing is considered to be the standard
method in nucleic acids studies, as it is able to directly identify
the specific mutations that may be present. However, it has the
disadvantages of high cost, long turnaround time and weak
sensitivity, limiting its practicality in diagnostic settings
(30). Therefore, the present
study attempted to perform HRM analysis, prior to sequencing, for
screening ABCB11 mutations in order to achieve high affectivity and
efficiency, as well as low cost.
HRM analysis is a powerful technique for the rapid
detection of mutations in double-stranded DNA samples that has the
potential to meet clinical demand (31). HRM involves precise monitoring of
the change in fluorescence caused by the release of a saturating
double-stranded DNA-binding fluorescent dye from a DNA duplex,
which is denatured by increasing temperature. Sequence variants,
including mutations and SNPs, are detected from the differences in
the melting profiles between the test and reference DNA (32).
In the present study, HRM analysis exhibited marked
affectivity and efficiency for detecting variants of the ABCB11
gene. It exhibited a high sensitivity (estimated 100% in this
study), high speed (30 min for processing 96 samples concurrently)
and low cost (the reagents used in the present study cost an
estimated US $0.5/sample/amplicon). Furthermore, the false-negative
rate was zero and the rate of false-positive calls, estimated to be
0.37%, was allowable as the mutations were further confirmed by
direct sequencing.
Mutation detection in the ABCB11 gene is important
in confirming the diagnosis of PFIC2. Among the seven mutations
identified in the study, p.Y337H, p.R696W, p.Q931P, p.D1131V and
p.H1198R are novel mutations according to data from The Human Gene
Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php). Also, none of
the mutations were detected in the control subjects. The missense
mutation p.Y337H (c.1009T>C) occurred in the transmembrane
domain (TMD) that led to an amino acid substitution from tyrosine
to histidine at position 337 in BSEP. By comparing amino acids at
this locus between species, tyrosine was evolutionarily conserved.
While the TMDs form the pore and define substrate specificity
(33), changes in TMDs impairs the
binding of substrate. Therefore, the change may affect the protein
features. Furthermore, the nucleotide change in ABCB11
(c.1009T>C) introduced a serine/arginine-rich splicing factor 6
(SRSF6) binding site, that has a role in constitutive splicing and
modulates the selection of alternative splice sites according to
the results of the ESEfinder3.0 software (http://rulai.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi).
In addition, precursor mRNA (pre-mRNA) splicing may be disturbed
(14). All of the other four
mutations occurred in intracellular loops. These mutations were
evolutionarily conserved and an comprehensive analysis of the
results of SIFT, PolyPhen-2, SNPs&GO revealed that they may be
deleterious with the exception of p.Q931P. Correlation with
clinical and genetic findings indicated that these mutations may be
functionally relevant. The missense mutation p.Y472C and nonsense
mutation p.R928X, have been reported in PFIC2 patients of European
populations and immunohistochemical staining for BSEP was
undetectable (26–28,34).
The previous studies are consistent with the present study, which
demonstrates that comprehensive analysis of functional prediction
is essential and useful. Although studies have demonstrated that
missense mutations in ABCB11 may impair BSEP processing and
function, or disrupt pre-mRNA splicing in vitro (14), further studies are required to
elucidate their definite roles.
ABCB11 gene mutations in children have been
identified in Asian populations, including in Thailand (35), India (36), Japan (37–39),
Taiwan (16,40,41)
and mainland China (17,42), the majority of which are different
from those reported in other populations. Common mutations that
have been reported in European populations, such as E297G and
D482G, were not detected in Chinese subjects. Therefore, mutations
in ABCB11 may be ethnicity-specific as hypothesized by
Ananthanarayanan and Li (43).
V444A and A1028A are two highly prevalent
polymorphisms. The allele frequency of V444A and A1028A has been
reported in Japanese and Caucasian populations (44). According to the present study, the
allele frequencies of V444A and A1028A were 74.5% and 67.2%,
respectively, in mainland Chinese populations. V444A has previously
been implicated in ICP and DIC with a higher allele frequency than
normal controls suggesting that this polymorphism may become
disease relevant in certain conditions, such as pregnancy and the
use of ethinylestradiol and levonorgestrel (11,45).
A1028A occurs in the TMD and it has been demonstrated that it
caused severe exon skipping (14).
However, as the number of patients tested in the present study is
small, it is not conclusive whether V444A or A1028A has a role in
PFIC2 or cholestasis. Further larger scale studies focusing on
specific populations are warranted to fully elucidate their roles.
As the frequency of the two SNPs is so high, sequencing directly
for exon 13 and exon 24 in case of omission is recommended.
In conclusion, the present study has established a
rapid genetic test that screens mutations and SNPs across the
entire ABCB11 coding regions. No common mutations in the ABCB11
gene were found in the Chinese population. V444A and A1028A were
two highly prevalent SNPs found in ABCB11 exons in the study
population; however, whether they are associated with pediatric
cholestatic diseases remains unclear. Further studies investigating
these mutations may enrich the knowledge of the ABCB11 gene and its
product, BSEP, and may therefore be beneficial to the personalized
management of individual patients in the future.
Acknowledgements
This study was sponsored by the Nanjing Municipal
Health Bureau fund and by the Department of Medical Genetics and
Jiangsu Key Laboratory of Molecular Medicine in Nanjing University
(Nanjing, China). The authors would like to thank their laboratory
for their technical assistance in the study.
Abbreviations:
|
PFIC2
|
progressive familial intrahepatic
cholestasis type 2
|
|
HRM
|
high-resolution melting curves
|
|
SNPs
|
single nucleotide polymorphisms
|
|
BSEP
|
bile salt export pump
|
|
GGT
|
gamma-glutamyltransferase
|
References
|
1
|
Keitel V, Burdelski M, Vojnisek Z, Schmitt
L, Häussinger D and Kubitz R: De novo bile salt transporter
antibodies as a possible cause of recurrent graft failure after
liver transplantation: a novel mechanism of cholestasis.
Hepatology. 50:510–517. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Morotti RA, Suchy FJ and Magid MS:
Progressive familial intrahepatic cholestasis (PFIC) type 1, 2, and
3: a review of the liver pathology findings. Semin Liver Dis.
31:3–10. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Harris MJ, Le Couteur DG and Arias IM:
Progressive familial intrahepatic cholestasis: genetic disorders of
biliary transporters. J Gastroenterol Hepatol. 20:807–817. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hori T, Nguyen JH and Uemoto S:
Progressive familial intrahepatic cholestasis. Hepatobiliary
Pancreat Dis Int. 9:570–578. 2010.
|
|
5
|
Nicolaou M, Andress EJ, Zolnerciks JK,
Dixon PH, Williamson C and Linton KJ: Canalicular ABC transporters
and liver disease. J Pathol. 226:300–315. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jankowska I and Socha P: Progressive
familial intrahepatic cholestasis and inborn errors of bile acid
synthesis. Clin Res Hepatol Gastroenterol. 36:271–274. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Strautnieks SS, Bull LN, Knisely AS, et
al: A gene encoding a liver-specific ABC transporter is mutated in
progressive familial intrahepatic cholestasis. Nat Genet.
20:233–238. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
van Mil SW, van der Woerd WL, van der
Brugge G, et al: Benign recurrent intrahepatic cholestasis type 2
is caused by mutations in ABCB11. Gastroenterology. 127:379–384.
2004.PubMed/NCBI
|
|
9
|
Andrade RJ, Robles M, Ulzurrun E and
Lucena MI: Drug-induced liver injury: insights from genetic
studies. Pharmacogenomics. 10:1467–1487. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dixon PH, van Mil SW, Chambers J, et al:
Contribution of variant alleles of ABCB11 to susceptibility to
intrahepatic cholestasis of pregnancy. Gut. 58:537–544. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Meier Y, Zodan T, Lang C, et al: Increased
susceptibility for intrahepatic cholestasis of pregnancy and
contraceptive-induced cholestasis in carriers of the 1331T>C
polymorphism in the bile salt export pump. World J Gastroenterol.
14:38–45. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Stieger B and Geier A: Genetic variations
of bile salt transporters as predisposing factors for drug-induced
cholestasis, intrahepatic cholestasis of pregnancy and therapeutic
response of viral hepatitis. Expert Opin Drug Metab Toxicol.
7:411–425. 2011. View Article : Google Scholar
|
|
13
|
Müllenbach R, Weber SN, Krawczyk M, et al:
A frequent variant in the human bile salt export pump gene ABCB11
is associated with hepatitis C virus infection, but not liver
stiffness in a German population. BMC Gastroenterol. 12:632012.
|
|
14
|
Byrne JA, Strautnieks SS, Ihrke G, et al:
Missense mutations and single nucleotide polymorphisms in ABCB11
impair bile salt export pump processing and function or disrupt
pre-messenger RNA splicing. Hepatology. 49:553–567. 2009.
View Article : Google Scholar
|
|
15
|
Pauli-Magnus C, Kerb R, Fattinger K, et
al: BSEP and MDR3 haplotype structure in healthy Caucasians,
primary biliary cirrhosis and primary sclerosing cholangitis.
Hepatology. 39:779–791. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen HL, Liu YJ, Su YN, et al: Diagnosis
of BSEP/ABCB11 mutations in Asian patients with cholestasis using
denaturing high performance liquid chromatography. J Pediatr.
153:825–832. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu LY, Wang ZL, Wang XH, Zhu QR and Wang
JS: ABCB11 gene mutations in Chinese children with progressive
intrahepatic cholestasis and low gamma glutamyltransferase. Liver
Int. 30:809–815. 2010. View Article : Google Scholar
|
|
18
|
Nguyen-Dumont T, Calvez-Kelm FL, Forey N,
et al: Description and validation of high-throughput simultaneous
genotyping and mutation scanning by high-resolution melting curve
analysis. Hum Mutat. 30:884–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen HL, Liu YJ, Su YN, et al: Diagnosis
of BSEP/ABCB11 mutations in Asian patients with cholestasis using
denaturing high performance liquid chromatograph. J Pediatr.
153:825–832. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Schwarz JM, Rödelsperger C, Schuelke M and
Seelow D: MutationTaster evaluates disease-causing potential of
sequence alterations. Nat Methods. 7:575–576. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ng PC and Henikoff S: SIFT: Predicting
amino acid changes that affect protein function. Nucleic Acids Res.
31:3812–3814. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Castellana S and Mazza T: Congruency in
the prediction of pathogenic missense mutations: state-of-the-art
web-based tools. Brief Bioinform. 14:448–459. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Adzhubei I, Jordan DM and Sunyaev SR:
2013.Predicting Functional Effect of Human Missense Mutations Using
PolyPhen-2. Curr Protocols in Human Genetics. 76:7.20.1–7.20.41
|
|
24
|
Calabrese R, Capriotti E, Fariselli P,
Martelli PL and Casadio R: Functional annotations improve the
predictive score of human disease-related mutations in proteins.
Hum Mutat. 30:1237–1244. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Leabman MK, Huang CC, DeYoung J, et al:
Natural variation in human membrane transporter genes reveals
evolutionary and functional constraints. Proc Natl Acad Sci USA.
100:5896–5901. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Evason K, Bove KE, Finegold MJ, et al:
Morphologic findings in progressive familial intrahepatic
cholestasis 2 (PFIC2): correlation with genetic and
immunohistochemical studies. Am J Surg Pathol. 35:687–696. 2011.
View Article : Google Scholar
|
|
27
|
Strautnieks SS, Byrne JA, Pawlikowska L,
et al: Severe bile salt export pump deficiency: 82 different ABCB11
mutations in 109 families. Gastroenterology. 134:1203–1214. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu C, Aronow BJ, Jegga AG, et al: Novel
resequencing chip customized to diagnose mutations in patients with
inherited syndromes of intrahepatic cholestasis. Gastroenterology.
132:119–126. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Acalovschi M, Tirziu S, Chiorean E,
Krawczyk M, Grünhage F and Lammert F: Common variants of ABCB4 and
ABCB11 and plasma lipid levels: a study in sib pairs with
gallstones, and controls. Lipids. 44:521–526. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wheeler DA, Srinivasan M, Egholm M, et al:
The complete genome of an individual by massively parallel DNA
sequencing. Nature. 452:872–876. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
van der Stoep N, van Paridon CD, Janssens
T, et al: Diagnostic guidelines for high-resolution melting curve
(HRM) analysis: an interlaboratory validation of BRCA1 mutation
scanning using the 96-well LightScanner. Hum Mutat. 30:899–909.
2009.PubMed/NCBI
|
|
32
|
van der Stoep N, van Paridon CD, Janssens
T, et al: Diagnostic guidelines for high-resolution melting curve
(HRM) analysis: an interlaboratory validation of BRCA1 mutation
scanning using the 96-well LightScanner. Hum Mutat. 30:899–909.
2009.PubMed/NCBI
|
|
33
|
Zolnerciks JK, Andress EJ, Nicolaou M and
Linton KJ: Structure of ABC transporters. Essays Biochem. 50:43–61.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Matte U, Mourya R, Miethke A, et al:
Analysis of gene mutations in children with cholestasis of
undefined etiology. J Pediatr Gastroenterol Nutr. 51:488–493. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Treepongkaruna S, Gaensan A, Pienvichit P,
et al: Novel ABCB11 mutations in a Thai infant with progressive
familial intrahepatic cholestasis. World J Gastroenterol.
15:4339–4342. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nobili V, Di Giandomenico S, Francalanci
P, Callea F, Marcellini M and Santorelli FM: A new ABCB11 mutation
in two Italian children with familial intrahepatic cholestasis. J
Gastroenterol. 41:598–603. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Goto K, Sugiyama K, Sugiura T, et al: Bile
salt export pump gene mutations in two Japanese patients with
progressive familial intrahepatic cholestasis. J Pediatr
Gastroenterol Nutr. 36:647–650. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Saito S, Iida A, Sekine A, et al: Three
hundred twenty-six genetic variations in genes encoding nine
members of ATP-binding cassette, subfamily B (ABCB/MDR/TAP), in the
Japanese population. J Hum Genet. 47:38–50. 2002. View Article : Google Scholar
|
|
39
|
Kim SR, Saito Y, Itoda M, et al: Genetic
variations of the ABC transporter gene ABCB11 encoding the human
bile salt export pump (BSEP) in a Japanese population. Drug Metab
Pharmacokinet. 24:277–281. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen HL, Chang PS, Hsu HC, et al: FIC1 and
BSEP defects in Taiwanese patients with chronic intrahepatic
cholestasis with low gamma-glutamyltranspeptidase levels. J
Pediatr. 140:119–124. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen ST, Chen HL, Su YN, et al: Prenatal
diagnosis of progressive familial intrahepatic cholestasis type 2.
J Gastroenterol Hepatol. 23:1390–1393. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu LY, Wang XH, Lu Y, Zhu QR and Wang JS:
Association of variants of ABCB11 with transient neonatal
cholestasis. Pediatr Int. 55:138–144. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ananthanarayanan M and Li Y: PFIC2 and
ethnicity-specific bile salt export pump (BSEP, ABCB11) mutations:
where do we go from here? Liver Int. 30:777–779. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lang T, Haberl M, Jung D, et al: Genetic
variability, haplotype structures, and ethnic diversity of hepatic
transporters MDR3 (ABCB4) and bile salt export pump (ABCB11). Drug
Metab Dispos. 34:1582–1599. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lang C, Meier Y, Stieger B, et al:
Mutations and polymorphisms in the bile salt export pump and the
multidrug resistance protein 3 associated with drug-induced liver
injury. Pharmacogenet Genomics. 17:47–60. 2007. View Article : Google Scholar : PubMed/NCBI
|