Introduction
Pulmonary arterial hypertension (PAH), a group of
diseases defined as a mean pulmonary arterial pressure of >25
mmHg at rest with an pulmonary arterial wedge pressure of <15
mmHg, occurs with an idiopathic aetiology or in association with
diseases including congenital heart malformation, connective tissue
disease or human immunodeficiency virus infection (1). PAH is essentially a vasculopathy in
the pulmonary circulation, manifesting narrowing or occlusion of
the vessel lumen in pre-capillary arterioles due to the formation
of neointimal or plexiform lesions. Despite extensive evidence for
the mechanisms underlying the pathophysiological etiology of PAH,
few effective therapeutic modalities have emerged as a result
(2,3).
Dichloroacetate (DCA) is as a metabolism modulator,
which has been utilized for approximately three decades as the
first line drug in the treatment of diseases associated with
mitochondrial dysfunction, including congenital lactic acidosis
(4). Several previous studies
indicated that DCA may reverse the medial thickening in the
monocrotaline (MCT) model, hypoxic PAH in rats and SM22α-targeted
over-expression of the serotonin transporter in mouse models
(5–7). The mechanism underlying this effect
may be the inactivation of pyruvate dehydrogenase (PDH) kinase and
transportation of pyruvate into the Krebs cycle instead of lactic
production, which leads to depolarization of the mitochondrial
membrane potential and initiation of apoptosis in pulmonary
arterial smooth muscle cells (SMCs) (8). However, the efficacy of DCA in
MCT-treated pneumonectomized rats is yet to be investigated. DCA
was identified to be effective in promoting apoptosis in a variety
of cancer cell lines (9,10). Furthermore, the treatment of
epithelial ovarian cancer cells with DCA markedly increased
superoxide dismutase (SOD) transcription and protein expression
levels and ultimately resulted in the inactivation of
hypoxia-inducible factor-1α (HIF-1α) and activation of the
mitochondrial apoptosis pathway (11).
Hypoxia-inducible factor-1 (HIF-1), a heterodimer of
HIF-1α and HIF-1β, regulates the transcriptional activation of
genes involved in energy metabolism, vasomotor tone and
angiogenesis, as part of an adaptive molecular response to low
oxygen availability (12). Several
previous studies suggested that HIF-1α activation was involved in
angiogenesis in patients with PAH complicated with congenital heart
malformation or idiopathic forms other than hypoxia-induced PAH
(13). Consistent with this, a
previous study by our group demonstrated that HIF-1α levels were
markedly increased in the neointimal lesion areas in
pneumonectomized rats following insult with MCT (14). Accumulating evidence has indicated
that growth factors, thrombin, angiotensin II, cytokines,
transforming growth factor-β or oxidative stress may account for
HIF-1α activation in PAH under non-hypoxic conditions (15). Of note, numerous of these
aforementioned factors also stimulated the generation of reactive
oxygen species (ROS), whereas inhibition of ROS formation decreased
HIF-1α protein levels, suggesting that ROS may have an important
role in the regulation of HIF-1α (16,17).
Therefore, it is hypothesized that DCA is effective
in preventing or reversing intimal lesions by inhibition of
ROS-induced HIF-1α activation, which is mediated by upregulation or
activation of SOD. In the present study, the effect of DCA on the
pulmonary vascular remodeling in rats was examined, utilizing two
regimens, consisting of a preventive or late intervention protocol.
Furthermore, the HIF-1α and ROS expression levels and SOD activity
were detected and the possible mechanisms underlying DCA-induced
amelioration of PAH were also investigated.
Materials and methods
Experimental animals
Healthy male Sprague-Dawley rats (nine weeks old)
were purchased from Xipuer-Bikai Laboratory Animal Co., Ltd.
(Shanghai, China) and housed in dry-raising cages (n=6/cage) at the
animal center of the Affiliated Hospital of Nanjing University
Medical School (Nanjing, China) in a standard 12-h reverse
day/night cycle at an ambient temperature of 26°C. A diet of stock
laboratory diet and tap water were available ad libitum.
They were allowed a recovery period of one week prior to the
experimental procedures. All experiments were performed with
approval from the Committee of Nanjing University Medical School
for Laboratory Animal Care and Use (Nanjing, China).
Left pneumonectomy
On day one, the rats were anesthetized with ketamine
(150 mg/kg, intraperitoneal injection) and orally intubated. Then,
the rats underwent left pneumonectomy via left thoracotomy, as
previously described (14).
MCT administration
MCT (Sigma-Aldrich, St. Louis, MO, USA) was
dissolved in dimethyl sulfoxide (DMSO) at a concentration of 30
mg/ml. On day eight, the rats were injected subcutaneously in the
cervical areas with MCT (2 ml/kg) as previously described (14).
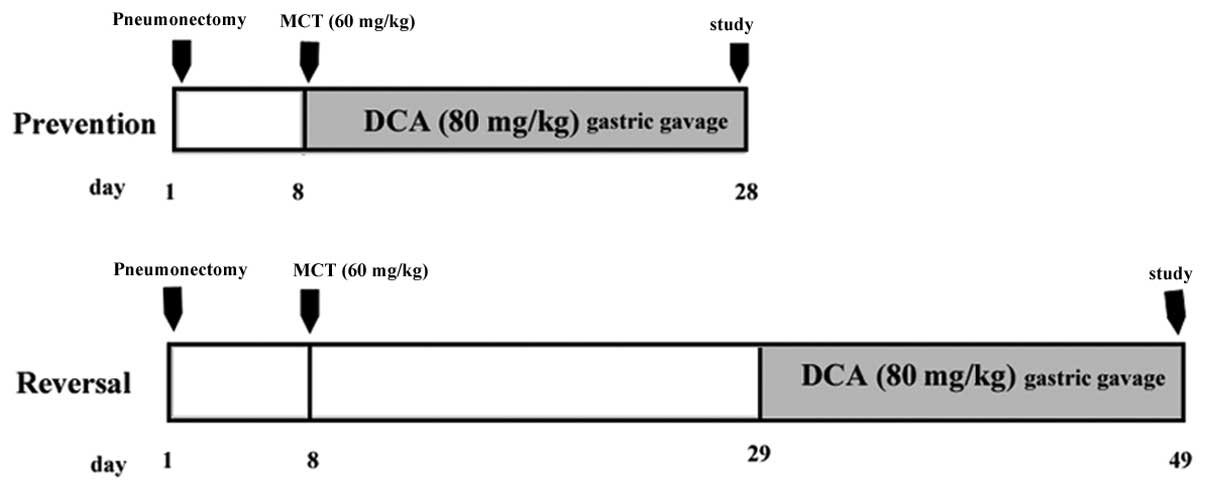
Experimental groups
Prevention study
Four groups of male Sprague-Dawley rats (weighing,
200–250 g; n=8/group) were used in the present study. Two groups of
rats undergoing left unilateral pneumonectomized were injected with
MCT (60 mg/kg, subcutaneous injection) seven days following surgery
and the other two groups received sham surgery and DMSO injections.
On day eight, one of the MCT- and DMSO-injected groups received 12
g/l DCA (Sigma-Aldrich) gastric gavages at a dose of 80 mg/kg and
the other two groups received the vehicle (normal saline) daily. On
day 28, hemodynamic measurements were performed in the anesthetized
rats.
Reversal study
The 32 male Sprague-Dawley rats were randomly
divided into four groups as described above. On day 29, one of the
MCT- and DMSO-injected groups received 12 g/l DCA gastric gavages
at a dose of 80 mg/kg, whereas the other two groups received the
vehicle (normal saline). Following 20 days of treatment,
hemodynamic measurements and morphometric analyses were performed.
The administration in the different groups is presented as a
schematic in Fig. 1.
Hemodynamic studies and tissue
preparation
At the end of the study as indicated above, the rats
were anesthetized with ketamine (150 mg/kg, intraperitoneal
injection). Their tracheas were orally intubated with a 16-gauge
intravenous catheter and mechanical ventilation commenced using a
rodent respirator (tidal volume, 8 ml/kg; respiratory rate, 60
min−1). The rats’ right carotid artery was cannulated
with a 20-gauge intravenous catheter to record systemic blood
pressure. Pulmonary hemodynamic parameters were measured in the
open chest via direct puncture of the right ventricle outflow tract
and then advancing the catheter into the pulmonary artery trunk
(also confirmed by standard right ventricle or pulmonary arterial
pressure trace on the monitor screen) connected to the pressure
transducer of an Eagle400 monitor (General Electric Company,
Fairfield, CT, USA). The right ventricle systolic pressure (RVSP),
mean pulmonary arterial pressure (PAP) and mean carotid blood
pressure (ABP) was recorded following 1 min of stabilization. All
rats were then euthanized by exsanguination, and the right lungs
(for histology and morphometry) and the hearts [for right ventricle
(RV)-to-left ventricle (LV) + intra-ventricle septum (S) ratio
(RV/LV+IVS)] were collected. The right low lobe of lung was
dissected, snap-frozen and maintained at a temperature of −80°C
until analyzed.
Morphological analysis
Histological changes of the pulmonary arteries (PAs)
were quantified by morphometry as described below. Isolated lungs
were inflated via the trachea with 10% formalin solution at 20 cm
H2O pressure and fixed in 10% formalin. Paraffin
sections of 5 μm were cut and stained with Elastin van Gieson and
assessed microscopically to determine the degree of arterial wall
thickness. In each lung section, ten small pulmonary arteries (PAs;
50–100 μm in diameter) were analyzed at a magnification of ×400 in
a blind manner. Medial wall thickness was expressed as the
summation of two points of medial thickness/external diameter × 100
(%). Intra-acinar (precapillary) PAs (25 vessels each) were
assessed for occlusive lesions as grade 0 for no evidence of
neointima lesion, grade 1 for <50% luminal occlusion and grade 2
for >50% luminal occlusion. There was no evidence of neointimal
lesion formation in any PAs from normal rats (all PAs were graded
as 0).
Immunohistochemistry
Slides were quenched in 3% hydrogen peroxide and the
antigen was retrieved using a pressure cooker in 10 mM citrate
buffer at pH 6. The slides were blocked in serum and the primary
antibody was applied overnight at 4°C. Immunodetection of HIF-1α
(dilution, 1:100; rabbit polyclonal; Novus Biologicals, Littleton,
CO, USA) was performed with horseradish peroxidase-conjugated
anti-rabbit secondary antibody (KPL, Inc., Gaithersburg, MD, USA)
and DAB stain (Dako, Glostrup, Denmark). The slides were
hematoxylin counterstained. The primary antibody (Dako Denmark A/S,
Glostrup, Denmark) was omitted in the negative control for every
group of slides.
Immunoblot assay
Whole cell, nuclear extracts were prepared according
to manufacturer’s instructions using the Nuclear Extract kit
(Nanjing KeyGen Biotech. Co., Ltd., Nanjing, China). Whole cell
extracts were prepared by lysing lung tissue in
radioimmunoprecipitation assay buffer (protein extraction reagent
with protease inhibitor cocktail, PMSF and phosphatase cocktail;
Nanjing KeyGen Biotech., Co., Ltd., Nanjing, China) for 20 min on
ice. Lysates were centrifuged (4°C) at 16,000 × g for 10 min and
the supernatant was collected for western blot analysis. Protein
concentration was determined using the Bradford assay (Nanjing
KeyGen Biotech. Co., Ltd.).
Western blot analysis
Western blots were performed by purifying 75 μg of
nuclear or whole cell protein extracts by 10% SDS-PAGE and
transferring the purified protein to polyvinyl difluoride membranes
by standard procedures. Membranes were blocked with tris-buffered
saline/Tween-20 with 10% non-fat milk for 2 h and then either
incubated overnight at 4°C or 4 h at room temperature with the
respective primary antibody, including HIF-1α (dilution, 1:500;
Novus Biologicals), Kv1.5 (dilution, 1:1,000; Abcam, Cambridge,
UK), survivin (1:1000 dilution; Abcam) and histone-3, GAPDH (each
dilution, 1:2,000; Cell Signaling Technology, Inc., MA, USA).
Immunoreactive bands were visualized using horseradish
peroxidase-conjugated anti-mouse or anti-rabbit secondary antibody
(dilution, 1:5,000; Upstate Biotechnology, Billerica, MA, USA) and
enhanced chemiluminescence reagent (Western chemiluminescent
detection system; Pierce Biotechnology, Inc., Rockford, IL, USA).
Relative immunoreactive levels of proteins were quantified using
the ChemiDoc XRS imaging system and Quantity One software (version
4.5; Bio-Rad, Hercules, CA, USA).
Assessment of ROS and SOD
Detection of ROS
For the tissue homogenates, lung tissue was rinsed
with 1× phosphate-buffered saline (PBS) to remove excess blood,
homogenized in 20 ml 1× PBS and stored overnight at <−20°C.
Following the two freeze-thaw cycles that were performed to break
the cell membranes, the homogenates were centrifuged for 5 min at
5,000 × g. The supernatant was removed and assayed immediately or
aliquoted and stored at <−20°C. ROS was detected according to
the manufacturer’s instructions by using a rat ROS ELISA assay kit
(R&D Systems, Inc., Minneapolis, MN, USA).
SOD activity assay
Prior to the assays, tissue samples were thawed on
ice and homogenized (Polytron PT3100; Brinkman Instruments, Littau,
Switzerland) in selected buffers. The total superoxide dismutase
activities were measured by using a water-soluble formazan dye kit
(Dojindo Molecular Technologies, Inc., Shanghai, China) according
to the manufacturer’s instructions. To measure manganese SOD
(MnSOD) activity, copper/zinc SOD (Cu/Zn-SOD) activity was blocked
with 1 mmol/l potassium cyanide. The amount of protein was measured
using a bicinchoninic acid protein assay (Sigma-Aldrich). Enzymatic
activity was expressed in units/mg protein (U/mgprot).
Statistical analysis
Statistical analysis was performed with GraphPad
Prism 5 (GraphPad Software, San Diego, CA, USA). The results are
presented as the mean ± standard error of the mean. Analysis of
variance followed by Bonferroni’s post-hoc test to compare the mean
values among the various groups. A P-value of <0.05 was
considered to indicate a statistically significant difference.
Results
Effect of DCA on hemodynamic parameters
and the formation of neointimal lesions
Consistent with previous results, left unilateral
pneumonectomy plus MCT treatment caused a significant increase in
the mean PAP, RVSP and right ventricle hypertrophy in rats as
compared with the reference control rats on day 28 (mean PAP,
33±5.5 vs. 18±2.1 mmHg; RVSP, 52±5.3 vs. 23±2.2 mmHg; RV/LV+IVS,
43±3.3 vs. 28±2.2%; P<0.05). The preventive treatment with DCA
significantly repressed the elevation of pulmonary pressure (mean
PAP, 24±2.8 vs. 33±5.5 mmHg; RVSP, 31±3.2 vs. 52±5.3 mmHg;
P<0.05) and right ventricle hypertrophy (RV/LV+IVS, 29±2.8 vs.
43±3.3%) in pulmonary hypertensive rats (Figs. 2A and 3H).
 | Figure 2Effect of DCA on the hemodynamic
parameters in MCT-treated pneumonectomized rats. The MCT-treated
pneumonectomized rats received preventive (day 8–28) and delayed
(day 29–49) DCA gastric gavage treatment, respectively. At the end
of the study, pulmonary hemodynamic parameters (mPAP and RVSP) were
measured on (A) day 28 and (B) day 49, respectively. The data are
presented as the mean ± standard error of the mean (n=8).
*P<0.05 vs. reference control rats on day 28 or 49.
Con, rats subjected to sham surgery and DMSO and treated with
vehicle (saline); DCA, rats subjected to sham surgery and DMSO and
treated with DCA (80 mg/kg daily) gavage treatment;
PM28, rats subjected to pneumonectomy and MCT and
treated with vehicle from day 8–28; PMD8~28, rats
subjected to pneumonectomy and MCT and treated with DCA from day
8–28; PM49, rats subjected to pneumonectomy and MCT and
treated with vehicle from day 29–49; PMD29~49, rats
subjected to pneumonectomy and MCT and treated with DCA from day
29–49. mPAP, mean pulmonary arterial pressure; DCA,
dichloroacetate; MCT, monocrotaline; RVSP, right ventricular
systolic pressure; DMSO, dimethyl sulfoxide. |
 | Figure 3Effect of DCA on pulmonary arterial
medial wall thickness and RV/LV+IVS. The MCT-treated
pneumonectomized rats received preventive (day 8–28) and delayed
(day 29–49) DCA gastric gavage treatment, respectively. At the end
of the study, the right lung and heart were dissected for
determination of lung vascular injury and right ventricle
hypertrophy, respectively. Rat lung tissues were stained with
H&E. View of lung (magnification, ×10) from a rat subjected to
sham operation and DMSO and treated with (A) saline or (D) DCA
receiving vehicle from day 8–28. Rat subjected to pneumonectomy and
treated with MCT receiving (B) vehicle or (C) DCA gastric gavage
from day 8–28. Rat subjected to pneumonectomy and treated with MCT
receiving (E) vehicle or (F) DCA gastric gavage from day 29–49. The
vessels marked with black arrows exhibited medial hypertrophy and
luminal narrowing and white arrow heads indicating completed
obliteration of several small arteriole lumens. (H&E stain;
scale bar in A–F, 100 μm). (G) Effect of DCA treatment on the
percentage of medial thickness in pulmonary arterioles and (H)
right ventricle hypertrophy index. The data are presented as the
mean ± standard error of the mean (n=8). *P<0.05,
**P<0.01 vs. reference control rats.
#P<0.05, ##P<0.01 vs. MCT-treated
pneumonectomized rats receiving vehicle from day 8–28. Con, rats
subjected to sham surgery and DMSO and treated with vehicle
(saline); DCA, rats subjected to sham surgery and DMSO and treated
with DCA (80 mg/kg daily) gavage treatment; PM28, rats
subjected to pneumonectomy and MCT and treated with vehicle from
day 8–28; PMD8~28, rats subjected to pneumonectomy and
MCT treated with DCA from day 8–28; PM49, rats subjected
to pneumonectomy and MCT and treated with vehicle from day 29–49;
PMD29~49, rats subjected to pneumonectomy and MCT and
treated with DCA from day 29–49. DMSO, dimethyl sulfoxide; DCA,
dichloroacetate; MCT, monocrotaline; RV/LV+IVS, right
ventricle/left ventricle+ intra-ventricle septum; H&E,
hematoxylin and eosin. |
Of note, the MCT-treated pneumonectomized rats
exhibited severe vascular remodeling, including medial wall
hypertrophy and occlusive neointimal formation leading to partial,
even complete, obliteration of pulmonary arterioles on day 28
(Figs. 3B and 4A). In the pulmonary arteries (50~100 μm
in diameter), the percentage of medial wall thickness was markedly
increased following left pneumonectomy plus MCT insults from 15% in
the reference control rats to 78%, which was reduced by the
treatment with early intervention of DCA (51±8.1 vs. 78±12.0%;
P<0.05; Fig. 3G). Furthermore,
Fig. 4B and E reveal that the
percentage of grade 1 and 2 occlusion in pre-acinar arteries
accounted for 24 and 20% of all pulmonary arterioles measured,
respectively, following treatment with DCA, which represented a
marked alleviation in neointimal lesion formation as compared with
the MCT-treated pneumonectomized rats receiving normal saline from
8–28 days following pneumonectomy (grade 1 and 2 occlusion
accounting for 44 and 40%, respectively).
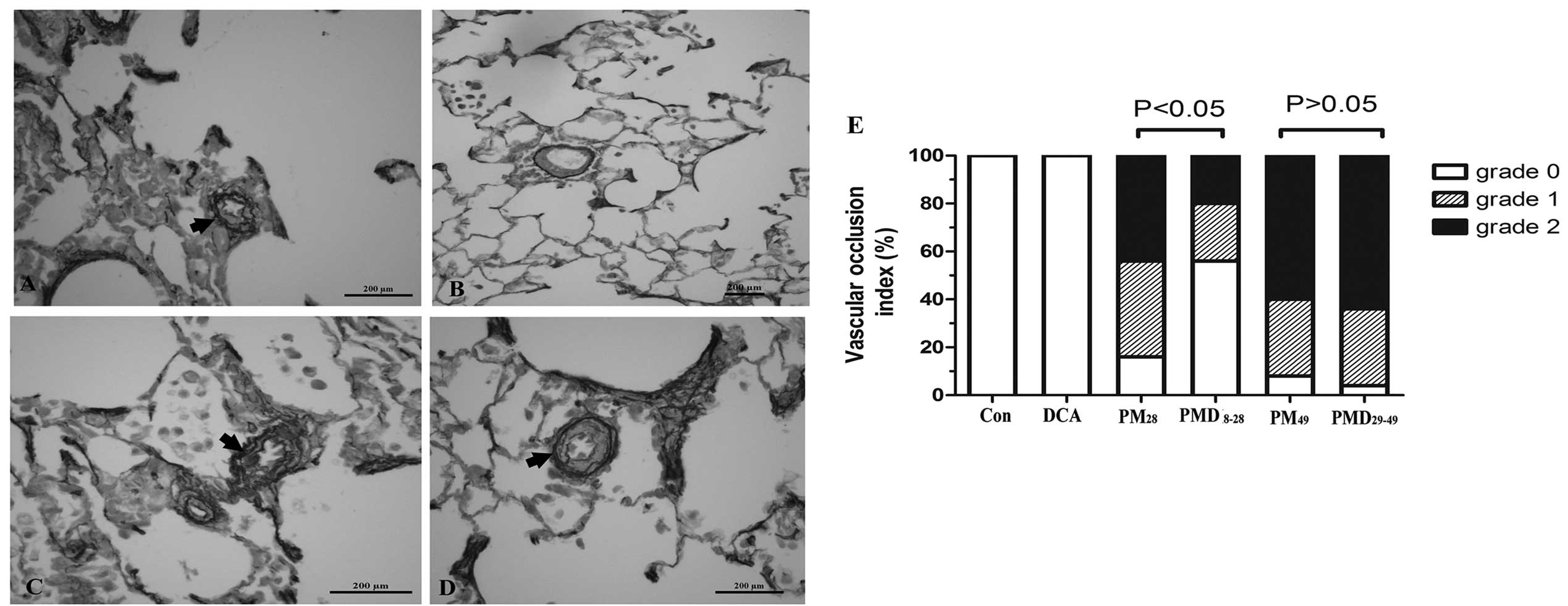 | Figure 4Effect of DCA on pulmonary arterioles
intimal lesion formation in rats treated with MCT following
pneumonectomy. The MCT-treated pneumonectomized rats received
preventive (day 8–28) and delayed (day 29–49) DCA gastric gavage
treatment, respectively. At the end of the experimental procedures,
the rat lung tissues were stained with Elastin van Gieson to
delineate the intima and media in pulmonary arterioles. View of
lung (magnification, ×40) from a rat subjected to pneumonectomy and
treated with MCT receiving (A) vehicle or (B) DCA gastric gavage
from day 8–28. Rats subjected to pneumonectomy and treated with MCT
receiving (C) vehicle or (D) DCA gastric gavage from day 29–49. The
vessels marked with arrows exhibited internal elastic laminal
interruption and neointimal formation, causing the narrowing of
pulmonary arteriole lumens. Scale bar in A–D, 200 μm. (E) Changes
of the vascular occlusion index of pulmonary arterioles following
preventive or delayed DCA treatment. Con, rats subjected to sham
surgery and DMSO and treated with vehicle (saline); DCA, rats
subjected to sham surgery and DMSO and treated with DCA;
PM28, rats subjected to pneumonectomy and MCT and
treated with vehicle from day 8–28; PMD8~28, rats
subjected to pneumonectomy and MCT and treated with DCA from day
8–28; PM49, rats subjected to pneumonectomy and MCT and
treated with vehicle from day 29–49; PMD29~49, rats
subjected to pneumonectomy and MCT and treated with DCA from day
29–49; DCA, dichloroacetate; MCT, monocrotaline; DMSO, dimethyl
sulfoxide. |
Compared with the MCT-treated pneumonectomized rats
on day 49, intervention with DCA from day 29–49 had little effect
on PAH (mean PAP, 35±1.9 vs. 36±3.5 mmHg; RVSP, 51±5.9 vs. 55±4.3
mmHg; P>0.05; Fig. 2B), RV
hypertrophy (RV/LV+IVS, 55±1.9% vs. 58±3.8%; P>0.05; Fig. 3H) and vascular remodeling (medial
wall thickness, 87±6.0 vs. 86±8.1%; grade 1 and 2 occlusion, 32 and
64 vs. 32% and 60%; P>0.05; Fig.
3G, 4C-E).
Effect of DCA on HIF-1α expression,
voltage-dependent potassium channel subtype 1.5 (Kv1.5) and
survivin expression
Fig. 5B and D show
intense staining of HIF-1α in occlusive neointimal lesions in
MCT-treated pneumonectomized rats on day 28 and 49, respectively.
The immunohistochemical and western blotting assays indicated that
the early treatment with DCA significantly inhibited HIF-1α
activation, as illustrated in Fig.
5C and Fig. 6A and B. By
contrast, the activation of HIF-1α was not inhibited by DCA
treatment from day 29–49 (Fig. 5E,
and Fig. 6A and B).
 | Figure 5Immunohistochemical staining of
HIF-1α in pulmonary arterioles. Lung tissue from (A) Con, (B)
PM28, (C) PMD8~28, (D) PM49 and (E)
PMD29~49. (magnification in A, ×10; magnification in
B–E, ×40; scale bar in A, 100 μm; scale bar in B–E, 50 μm). Con,
rats subjected to sham surgery and DMSO and treated with vehicle
(saline); PM28, rats subjected to pneumonectomy and MCT
and treated with vehicle from day 8–28; PMD8~28, rats
subjected to pneumonectomy and MCT and treated with DCA from day
8–28; PM49, rats subjected to pneumonectomy and MCT and
treated with vehicle from day 29–49; PMD29~49, rats
subjected to pneumonectomy and MCT and treated with DCA from day
29–49. HIF-1α, hypoxia-inducible factor-1α; DCA, dichloroacetate;
MCT, monocrotaline; DMSO, dimethyl sulfoxide. |
 | Figure 6Western blot analysis of expression
of HIF-1α, Kv1.5 and survivin in lung tissue. (A) Representative
alteration of HIF-1α expression in one of three independent
experiments with identical results. (B) Density ratio of HIF-1α to
Histone-3. (C) Representative alteration of Kv1.5 or survivin,
expression in one of three separate experiments with the identical
results. (D) Density ratio of Kv1.5 or survivin to GAPDH. The data
are presented as the mean ± standard error of the mean (n=3).
*P<0.05, **P<0.01 vs. Con;
#P<0.05 vs. PM28. Con, rats subjected to
sham surgery and DMSO and treated with vehicle (saline); DCA, rats
subjected to sham surgery and DMSO and treated with DCA (80 mg/kg
daily) gavage treatment; PM28, rats subjected to
pneumonectomy and MCT and treated with vehicle from day 8–28;
PMD8~28, rats subjected to pneumonectomy and MCT treated
with DCA from day 8–28; PM49, rats subjected to
pneumonectomy and MCT and treated with vehicle from day 29–49;
PMD29~49, rats subjected to pneumonectomy and MCT and
treated with DCA from day 29–49. Kv1.5, voltage-dependent potassium
channel subtype 1.5; HIF-1α, HIF-1α, hypoxia-inducible factor-1α;
MCT, monocrotaline; DCA, dichloroacetate; DMSO, dimethyl
sulfoxide. |
The findings from the present study demonstrated
that the reduced expression of Kv1.5 was restored by early
intervention with DCA in MCT-treated pneumonectomized rats.
However, when DCA treatment was delayed, there was evident
restoration of Kv1.5 expression on day 49 in rats (Fig. 6C and D).
There is considerable evidence that survivin is a
key anti-apoptotic factor responsible for cell overgrowth (18). In the present study, the expression
of survivin in the MCT-treated pneumonectomized rats was ~4-fold
higher as compared with the normal control rats, which was
abrogated with DCA treatment (P<0.05). As demonstrated in
Fig. 6E and F, however, the
expression of survivin was not significantly different between the
MCT-treated pneumonectomized rats receiving vehicle or DCA from day
29–49.
Effect of DCA on ROS content and the
expression of SOD in lung tissue
In agreement with the previous studies, the rats
that underwent pneumonectomy and treatment with MCT exhibited
higher ROS production compared with the rats subjected to sham
surgery plus DMSO treatment (106±4.7 vs. 56±10.2 U/mgprot;
P<0.05) on day 28. As demonstrated in Fig. 7A, the intervention with DCA
effectively suppressed the increase of ROS, which was reduced by
almost 25% as compared with the MCT-treated pneumonectomized rats
receiving vehicle on day 28. However, the MCT-treated
pneumonectomized rats treated with DCA from day 29–49 had a higher
ROS content as compared with those receiving vehicle treatment
(115±8.8 vs. 98±5.4 U/mgprot; P>0.05).
 | Figure 7Effect of DCA on ROS production and
SOD activation. The MCT-treated pneumonectomized rats received
early (day 8–28) and delayed (day 29–49) DCA gastric gavage
treatment, respectively. At the end of the study, the content of
(A) ROS, (B) total SOD, (C) MnSOD, (D) Cu/Zn SOD in rat lung tissue
was measured by a colorimetric assay. Data are presented as the
mean ± standard error of the mean (n=8). *P<0.05,
**P<0.01 vs. Con; #P<0.05 vs.
PM28. Con, rats subjected to sham surgery and DMSO and
treated with vehicle (saline); DCA, rats subjected to sham surgery
and DMSO and treated with DCA (80 mg/kg daily) gavage treatment;
PM28, rats subjected to pneumonectomy and MCT and
treated with vehicle from day 8–28; PMD8~28, rats
subjected to pneumonectomy and MCT and treated with DCA from day
8–28; PM49, rats subjected to pneumonectomy and MCT and
treated with vehicle from day 29–49; PMD29~49, rats
subjected to pneumonectomy and MCT and treated with DCA from day
29–49. ROS, reactive oxygen species; SOD, superoxide dismutase;
MCT, monocrotaline; DCA, dichloroacetate; MnSOD, manganese SOD;
Cu/Zn SOD, copper/zinc SOD; DMSO, dimethyl sulfoxide. |
As is well-established, SOD has a key role in the
degradation of ROS. The activity of total SOD, MnSOD and Cu/Zn SOD
was investigated in the present study. The total SOD activity was
significantly suppressed in MCT-treated pneumonectomized rats,
which was, in part, restored by early intervention with DCA (66±4.4
vs. 106±21.8 U/mgprot; P<0.05; Fig.
7B). The activity of MnSOD did not exhibit any significant
difference among the six groups, as illustrated in Fig. 7C. However, of note, the activity
changes of total SOD were in parallel with the alteration of Cu/Zn
SOD activity. Fig. 7D reveals that
the activity of Cu/Zn SOD was markedly suppressed in MCT-treated
pneumonectomized rats on day 28 as compared with that in the normal
control rats (53±2.7 vs. 96±19.5 U/mgprot; P<0.05), which was
reversed by intervention with DCA (88±4.2 vs. 53±2.7 U/mgprot;
P<0.05). However, late intervention with DCA had a weak effect
on Cu/Zn SOD activity (Fig. 7D).
Of note, the MCT-treated pneumonectomized rats on day 49 exhibited
an unexpected increase of Cu/Zn SOD activity as compared with the
MCT-treated pneumonectomized rats receiving the vehicle from day
8–28.
Discussion
The results of the present study indicated that DCA
was effective in preventing the formation of neointimal lesions in
pulmonary arterioles of MCT-induced pneumonectomized rats and
increases in PAP. This effect may be attributed to Cu/Zn SOD
upregulation, HIF-1α inactivation and subsequent cell apoptosis in
the neointimal lesion areas. By contrast, late intervention with
DCA demonstrated little efficacy in the amelioration of PAP and
severe structural destruction in pulmonary arterioles.
DCA has been widely employed in a number of diseases
associated with mitochondrial dysfunction for approximately three
decades. Recent studies have demonstrated the beneficial effect of
DCA in MCT or hypoxic PAH models (5,6).
However, these aforementioned rat models are limited by their
inability to reproduce the full spectrum of vascular pathological
injuries observed in lung specimens from PAH patients. In
particular, these models lack the characteristic neointimal
formation which causes narrowing, or even obliteration of vessel
lumens in pre-capillary vessels, as well as the significant
elevation in pulmonary vascular resistance (19). Intimal lesions are commonly
regarded as a type of pulmonary vascular pathology representing
severe injury and represent the irreversibility status during
pulmonary vascular remodeling (20). Therefore, it is highly important to
prevent or reverse the formation of neointimal lesions. The rats
undergoing left lung resection followed by MCT subcutaneous
injection developed severe PAH, accompanied with severe occlusive
neointimal formation in pulmonary arterioles. Since its first
introduction by Okada et al (21), this model has been applied to test
the effectiveness of a variety of drugs (simvastatin, triptolide,
etc.) in PAH (21–23). However, the efficacy of DCA in this
rat model, to the best of our knowledge, has not been investigated
to date.
DCA is a well-characterized inhibitor of the protein
kinase of pyruvate dehydrogenase (PDH). DCA-mediated inhibition of
PDH kinase renders the majority of PDH in the active form, which
triggers a switch in pyruvate metabolism towards glucose oxidation
to CO2 in the mitochondria (24). Early intervention with DCA in the
present study effectively attenuated neointimal formation in
pulmonary hypertensive rats. This effect was accompanied by the
suppression of HIF-1α activation and the induction of Kv1.5
expression in the neointimal lesions. By contrast, no amelioration
in pulmonary vascular remodeling was observed in the late
intervention group. Concomitantly, late treatment with DCA did not
reverse the alterations of this transcription factor and Kv1.5
protein expression in pulmonary hypertensive rats. These data
suggested that HIF-1α inactivation and upregulation of Kv1.5 may be
the key factors responsible for preventing neointimal formation in
MCT-induced pneumonectomized rats treated with DCA. Accumulating
evidence indicated that MnSOD (SOD2) converts intramitochondrial
superoxide to diffusible hydrogen peroxide, which serves as a
redox-signaling molecule, regulating pulmonary vascular tone and
structure through effects on Kv1.5 and transcription factors
(15). The expression of MnSOD has
been identified as reduced in idiopathic PAH patients and in
fawn-hooded rats that spontaneously develop PAH (25,26).
The reduced expression or inactivation of MnSOD creates a
pseudohypoxic redox state in PAH characterized by normoxic
decreases in ROS, a shift from oxidative to glycolytic metabolism
and HIF-1α activation (27). DCA
corrects the mitochondrial abnormalities in several experimental
models of PAH, causing a regression of remodeled pulmonary
vasculature. Inconsistent with previous results, in the present
study, HIF-1α inactivation was attributed to DCA-induced elevation
in the activity of Cu/Zn SOD (SOD1 and SOD3), while changes in
MnSOD levels were not statistically significant (P>0.05).
Experimental findings from Dorfmüller et al (28) indicated that increased oxidative
stress and severe vascular remodeling occurred in MCT-treated rats
following left pneumonectomy. In addition, administration of
antioxidants attenuated the development of PAH, which suggested
that pulmonary oxidative stress regulates the development of PAH
(29). In the present study, a
change with opposite trends was observed between ROS and total SOD
production in rat lung specimens 20 days following MCT treatment.
Early treatment with DCA effectively prevented excessive ROS
formation, which was mediated by an upregulation of Cu/Zn SOD
activity. SOD catalyzes the dismutation of superoxide into oxygen
and hydrogen peroxide, and elevated hydrogen peroxide and NO levels
are known to block the induction of HIF-1α-associated genes and the
accumulation of HIF-1α (30).
Therefore, it is hypothesized that DCA-induced elevation of Cu/Zn
SOD activity may be involved in the inactivation of HIF-1α in
neointimal lesion areas. In accordance with the results of the
present study, the findings from Saed et al (11) indicated that SOD3 was upregulated
in a dose-dependent manner at the transcriptional and protein
levels following DCA treatment, which induced apoptosis, reduced
myeloperoxidase activation and HIF-1α transcription in two
epithelial ovarian cancer cell lines. The protective effect of SOD3
has also been confirmed by a study demonstrating that the
overexpression of SOD3 in bleomycin induced pulmonary hypertension
in rats (31). By contrast, in
another study, enhanced hydrogen peroxide levels promoted
thrombin-induced HIF-1α upregulation, whereas degradation of
hydrogen peroxide with catalase prevented HIF-1α activation in
pulmonary arterial SMCs (32).
Therefore, the regulatory role of hydrogen peroxide in the
induction of HIF-1α activation remains elusive. A study by Görlach
et al (33) indicated that
higher concentrations of hydrogen peroxide were more inclined to
prevent HIF-1α accumulation, suggesting that only moderate changes
in the redox state are required to activate the HIF pathway. By
contrast, a substantial increase in hydrogen peroxide levels (as
they may be observed at conditions of oxidative stress) does
inactivate the HIF system. In addition, the present study did not
observe any significant changes in the activity and protein
expression of MnSOD (data not shown). The rodent model used in the
present study, which is characterized by enhanced oxidative stress
and inflammatory cell recruitment in lungs, may account for the
distinct roles of three types of SOD in HIF-1α suppression.
There are several possible explanations for the late
intervention with DCA not being effective in the PAH rats. Firstly,
20 days following MCT insults in pneumonectomized rats, the
occlusion of pulmonary arterioles was severe, which compromised the
trafficking and absorption of DCA at the sites of neointimal
lesions. Secondly, as is consistent with the results by Dorfmüller
et al (28), ROS in the
lung tissue were markedly increased following MCT treatment in
pneumonectomized rats. The early release of ROS and the occurrence
of damage to the adjacent tissues may have produced enough ROS to
injure their distant target (pulmonary arteries) and trigger a
self-enhancing pro-oxidative and pro-inflammatory process, which
may not be affected by any delayed ROS-dependent therapy (34).
In conclusion, the present study showed that HIF-1α
was upregulated in the neointimal lesion areas of pulmonary
arterioles in MCT-treated pneumonectomized rats, which accounts for
the occlusive neointimal formation and severe pulmonary
hypertension. The results demonstrated that DCA was effective in
the upregulation of Cu/Zn SOD activation leading to HIF-1α
inactivation in pulmonary vessels. However, late treatment with DCA
was not able to attenuate pulmonary vascular remodeling and
hemodynamics in this well established PAH model. Investigating
methods to improve DCA transportation to neointimal lesion areas
and inhibit the amplified oxidative stress may be a novel strategy
for the treatment of PAH in the future.
Acknowledgements
This study was supported by grants from National
Natural Science Foundation Committee of China (project no.
30901406); Young Health Talents Project from Nanjing Municipal
Health Bureau; Natural Science Foundation Committee of Jiangsu
Province (BK2012532) and Fundamental Research Fund for Central
University of Nanjing University, China.
Abbreviations:
|
Cu/Zn SOD
|
copper/zinc superoxide dismutase
|
|
DCA
|
dichloroacetate
|
|
DMSO
|
dimethyl sulfoxide
|
|
HIF-1α
|
hypoxia inducible factor α
|
|
MCT
|
monocrotaline
|
|
MnSOD
|
manganese superoxide dismutase
|
|
PAH
|
pulmonary arterial hypertension
|
|
PAP
|
pulmonary arterial pressure
|
|
PBS
|
phosphate-buffered saline
|
|
PDH
|
pyruvate dehydrogenase
|
|
ROS
|
reactive oxygen species
|
|
RV/LV+IVS
|
right ventricle/left
ventricle+intra-ventricle septum
|
|
RVSP
|
right ventricle systolic pressure
|
|
Kv1.5
|
voltage-dependent potassium channel
subtype 1.5
|
References
|
1
|
McLaughlin VV, Davis M and Cornwell W:
Pulmonary arterial hypertension. Curr Probl Cardiol. 36:461–517.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tuder RM: Pathology of pulmonary arterial
hypertension. Semin Respir Crit Care Med. 30:376–385. 2009.
View Article : Google Scholar
|
|
3
|
Stamm JA, Risbano MG and Mathier MA:
Overview of current therapeutic approaches for pulmonary
hypertension. Pulm Circ. 1:138–159. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stacpoole PW, Nagaraja NV and Hutson AD:
Efficacy of dichloroacetate as a lactate-lowering drug. J Clin
Pharmacol. 43:683–691. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McMurtry MS, Bonnet S, Wu X, Dyck JR,
Haromy A, Hashimoto K, et al: Dichloroacetate prevents and reverses
pulmonary hypertension by inducing pulmonary artery smooth muscle
cell apoptosis. Circ Res. 95:830–840. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Michelakis ED, McMurtry MS, Wu XC, Dyck
JR, Moudgil R, Hopkins TA, et al: Dichloroacetate, a metabolic
modulator, prevents and reverses chronic hypoxic pulmonary
hypertension in rats: role of increased expression and activity of
voltage-gated potassium channels. Circulation. 105:244–250. 2002.
View Article : Google Scholar
|
|
7
|
Guignabert C, Tu L, Izikki M, Dewachter L,
Zadigue P, Humbert M, et al: Dichloroacetate treatment partially
regresses established pulmonary hypertension in mice with
SM22alpha-targeted overexpression of the serotonin transporter.
FASEB J. 23:4135–4147. 2009. View Article : Google Scholar
|
|
8
|
Michelakis ED, Webster L and Mackey JR:
Dichloroacetate (DCA) as a potential metabolic-targeting therapy
for cancer. Br J Cancer. 99:989–994. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wong JY, Huggins GS, Debidda M, Munshi NC
and De Vivo I: Dichloroacetate induces apoptosis in endometrial
cancer cells. Gynecol Oncol. 109:394–402. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Washington JT and Quintyne NJ:
Dichloroacetate induces different rates of cell death in cancer and
noncancer cell lines in vitro. Tumori. 98:142–151. 2012.PubMed/NCBI
|
|
11
|
Saed GM, Fletcher NM, Jiang ZL, Abu-Soud
HM and Diamond MP: Dichloroacetate induces apoptosis of epithelial
ovarian cancer cells through a mechanism involving modulation of
oxidative stress. Reprod Sci. 18:1253–1261. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Semenza GL: Involvement of
hypoxia-inducible factor 1 in pulmonary pathophysiology. Chest.
128(Suppl): 592S–594S. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tuder RM, Chacon M, Alger L, Wang J,
Taraseviciene-Stewart L, Kasahara Y, et al: Expression of
angiogenesis-related molecules in plexiform lesions in severe
pulmonary hypertension: evidence for a process of disordered
angiogenesis. J Pathol. 195:367–374. 2001. View Article : Google Scholar
|
|
14
|
Yan J, Shen Y, Wang Y and Li BB: Increased
expression of hypoxia inducible factor-1α in proliferating
neointimal lesions in a rat model of pulmonary arterial
hypertension. Am J Med Sci. 345:121–128. 2013.
|
|
15
|
Archer SL, Gomberg-Maitland M, Maitland
ML, Rich S, Garcia JG and Weir EK: Mitochondrial metabolism, redox
signaling, and fusion: a mitochondria-ROS-HIF-1alpha-Kv1.5
O2-sensing pathway at the intersection of pulmonary hypertension
and cancer. Am J Physiol Heart Circ Physiol. 294:H570–H578. 2008.
View Article : Google Scholar
|
|
16
|
Déry MA, Michaud MD and Richard DE:
Hypoxia-inducible factor 1: regulation by hypoxic and non-hypoxic
activators. Int J Biochem Cell Biol. 37:535–540. 2005.PubMed/NCBI
|
|
17
|
Bonello S, Zähringer C, BelAiba RS,
Djordjevic T, Hess J, Michiels C, et al: Reactive oxygen species
activate the HIF-1alpha promoter via a functional NFkappaB site.
Arterioscler Thromb Vasc Biol. 27:755–761. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
McMurtry MS, Archer SL, Altieri DC, et al:
Gene therapy targeting survivin selectively induces pulmonary
vascular apoptosis and reverses pulmonary arterial hypertention. J
Clin Invest. 115:1479–1491. 2005. View
Article : Google Scholar
|
|
19
|
Stenmark KR, Meyrick B, Galie N, Mooi WJ
and McMurtry IF: Animal models of pulmonary arterial hypertension:
the hope for etiological discovery and pharmacological cure. Am J
Physiol Lung Cell Mol Physiol. 297:L1013–L1032. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sakao S, Tatsumi K and Voelkel NF:
Reversible or irreversible remodeling in pulmonary arterial
hypertension. Am J Respir Cell Mol Biol. 43:629–634. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Okada K, Tanaka Y, Bernstein M, Zhang W,
Patterson GA and Botney MD: Pulmonary hemodynamics modify the rat
pulmonary artery response to injury. A neointimal model of
pulmonary hypertension. Am J Pathol. 151:1019–1025. 1997.PubMed/NCBI
|
|
22
|
Nishimura T, Vaszar LT, Faul JL, Zhao G,
Berry GJ, Shi L, et al: Simvastatin rescues rats from fatal
pulmonary hypertension by inducing apoptosis of neointimal smooth
muscle cells. Circulation. 108:1640–1645. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Faul JL, Nishimura T, Berry GJ, Benson GV,
Pearl RG and Kao PN: Triptolide attenuates pulmonary arterial
hypertension and neointimal formation in rats. Am J Respir Crit
Care Med. 162:2252–2258. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Madhok BM, Yeluri S, Perry SL, Hughes TA
and Jayne DG: Dichloroacetate induces apoptosis and cell-cycle
arrest in colorectal cancer cells. Br J Cancer. 102:1746–1752.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bowers R, Cool C, Murphy RC, Tuder RM,
Hopken MW, Flores SC, et al: Oxidative stress in severe pulmonary
hypertension. Am J Respir Crit Care Med. 169:764–769. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bonnet S, Michelakis ED, Porter CJ,
Andrade-Navarro MA, Thébaud B, Haromy A, et al: An abnormal
mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway
disrupts oxygen sensing and triggers pulmonary arterial
hypertension in fawn hooded rats: similarities to human pulmonary
arterial hypertension. Circulation. 113:2630–2641. 2006. View Article : Google Scholar
|
|
27
|
Fijalkowska I, Xu W, Comhair SAA, Janocha
AJ, Mavrakis LA, Krishnamachary B, et al: Hypoxia
inducible-factor1alpha regulates the metabolic shift of pulmonary
hypertensive endothelial cells. Am J Pathol. 176:1130–1138. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dorfmüller P, Chaumais MC, Giannakouli M,
Durand-Gasselin I, Raymond N, Fadel E, et al: Increased oxidative
stress and severe arterial remodeling induced by permanent
high-flow challenge in experimental pulmonary hypertension. Respir
Res. 12:1192011.
|
|
29
|
Xu D, Guo H, Xu X, Lu Z, Fassett J, Hu X,
et al: Exacerbated pulmonary arterial hypertension and right
ventricular hypertrophy in animals with loss of function of
extracellular superoxide dismutase. Hypertension. 58:303–309. 2011.
View Article : Google Scholar
|
|
30
|
Brüne B and Zhou J: Nitric oxide and
superoxide: interference with hypoxic signaling. Cardiovasc Res.
75:275–282. 2007.PubMed/NCBI
|
|
31
|
Van Rheen Z, Fattman C, Domarski S, Majka
S, Klemm D, Stenmark KR, et al: Lung extracellular superoxide
dismutase overexpression lessens bleomycin-induced pulmonary
hypertension and vascular remodeling. Am J Respir Cell Mol Biol.
44:500–508. 2011.
|
|
32
|
BelAiba RS, Djordjevic T, Bonello S,
Flügel D, Hess J, Kietzmann T, et al: Redox-sensitive regulation of
the HIF pathway under non-hypoxic conditions in pulmonary artery
smooth muscle cells. Biol Chem. 385:249–257. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Görlach A, Diebold I, Schini-Kerth VB,
Berchner-Pfannschmidt U, Roth U, Brandes RP, et al: Thrombin
activates the hypoxia-inducible factor-1 signaling pathway in
vascular smooth muscle cells: Role of the p22(phox)-containing
NADPH oxidase. Circ Res. 89:47–54. 2001.PubMed/NCBI
|
|
34
|
Dahal BK, Kosanovic D, Kaulen C,
Cornitescu T, Savai R, Hoffmann J, et al: Involvement of mast cells
in monocrotaline-induced pulmonary hypertension in rats. Respir
Res. 12:602011. View Article : Google Scholar : PubMed/NCBI
|