Introduction
At present, cancer is regarded as a disease that is
caused by genetic and epigenetic alterations (1). Increasing evidence demonstrates that
chromatin-mediated changes induce cancer, including DNA
methylation, histone variants and miRNA variation (2). Once histone variants are replaced by
normal histones, the chromatin is changed. Subsequently, unfolded
and remodeled chromatin controls access to the transcription start
site, which regulates gene transcription (3). Additionally, the correlation between
these changes and cancer progression has been established (4). Histone variants affect stable gene
expression, which participates in tumor progression and
differentiation (5).
Histones, which are highly conserved in mammals, are
crucial in regulating nuclear activation via changing the chromatin
structure (6,7). Yet, the mechanisms by which they
change chromatin structure are less understood. H3 and H2A, two
major variants associated with several tissue-restricted proteins,
have been revealed to be involved in this process (8). In these variants, due to its
canonical counterpart, macroH2A demonstrates a different
characterization. MacroH2A, the only histone with a tripartite
structure consisting of an N-terminal histone-fold, contains an
unstructured linker domain and a unique C-terminal macro domain
(9,10). Due to its structure, macroH2A
demonstrates the most frequent alterations among all the histone
variants (11,12). Kapoor et al (13) demonstrated that the loss of
macroH2A is associated with chromatin condensation and regulated
gene expression during melanoma developmental programs.
Furthermore, by knocking down macroH2A in melanoma cells, it was
confirmed that the loss of macroH2A promoted cancer development via
the transcriptional upregulation of CDK8. CDK8 is a member of the
cyclin-dependent protein kinase (CDK) family, which has regulatory
functions in the cell cycle (14,15).
This finding suggests that macroH2A affects melanoma progression
through changes in the cell cycle. However, to date, information is
lacking regarding the effects of macroH2A on cell cycle regulatory
genes.
The cell cycle involves strict events that control
cell division and proliferation. The key factors, including CDKs
and cyclins, trigger the transition in the process of the cell
cycle (16–18). During this event, dysfunctional
expression of genes induced by normal metabolic activity or
environmental factors arrested or delayed checkpoints prior to cell
division (19,20). In addition, CDK/cyclin B is
important in stabilizing the genome in S phase (21,22).
Thus, abnormal expression of these factors disorganized the process
of cell division (23,24).
In the present study, the effects of macroH2A on
cell cycle-related genes were examined in melanoma. MacroH2A
interference, overexpression, overexpression rescue and
interference rescue treatments were performed by the transfection
of short hairpin (sh)RNA vectors and/or overexpression vectors.
Following transfection, macroH2A expression was determined. By
employing flow cytometry, the regulation of the B16 melanoma cell
line proliferation by macroH2A was confirmed. Furthermore, the
regulation of macroH2A on cyclin D and CDK were analyzed in the B16
melanoma cell line. The present study provides insights into the
effects of macroH2A on melanoma progression.
Materials and methods
Ethics statement
The present study obtained ethics approval from the
ethics committee at Xiangya Hospital, Central South University
(Changsha, China). The storage of samples for exploratory
immunological analyses was also ethically approved.
Cell culture
The B16 mouse melanoma cells were obtained from the
American Type Culture Collection (Manassas, VA, USA). The cells
were grown in RPMI-1640 medium supplemented with 10% fetal bovine
serum (Invitrogen Life Technologies, Carlsbad, CA, USA) in 5%
CO2 and saturated humidity at 37°C. Cell viability was
estimated by trypan blue exclusion. For cell morphology
examination, cells were grown on a chambered coverglass system
(Thermo Scientific, Rockford, IL, USA) and observed with an
inverted microscope (Nikon Corporation, Tokyo, Japan).
Vector design and transfection
For the comparison of biophysical properties
following knockdown and overexpression of macroH2A, the macroH2A
interference and overexpression vectors were constructed. The
recombinant expression plasmid (macroH2A shRNA plasmid) was
purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA;
sc-62576-SH). The recombinant expression plasmid expressing
macroH2A was constructed. Briefly, the open reading frame of
macroH2A (GenBank accession no. NM012015) was cloned into the
pcDNA3.1(t) plasmid (Invitrogen Life Technologies) between
XhoI and BamHI sites to build recombinant pc3.1-h
macroH2A plasmids. The cells were transfected with
pcDNA3.1(t)-macroH2A and/or the macroH2A shRNA plasmid using
Lipofectamine™ 2000 (Invitrogen Life Technologies) according to the
manufacturer’s instructions. Following 24 h of transfection, the
cells were harvested and used for the following experiments.
The cells were randomly divided into five groups
(five parallel treatments per group), including the control group
(non-treated group), macroH2A interference group (1 μg macroH2A
shRNA plasmid transfection), macroH2A overexpression group (1 μg
pcDNA3.1(t)-macroH2A plasmid transfection), macroH2A overexpression
rescue group (1 μg pcDNA3.1(t)-macroH2A plasmid transfection for 12
h following 1 μg macroH2A shRNA plasmid transfection for 12 h) and
the macroH2A interference rescue group (1 μg macroH2A shRNA plasmid
transfection for 12 h following 1 μg pcDNA3.1(t)-macroH2A plasmid
transfection for 12 h).
Quantitative polymerase chain reaction
(qPCR)
In order to analyze mRNA expression among different
groups, qPCR was performed. All the primers and probes were
designed by Applied Biosystems (Foster City, CA, USA), which
hybridized between exons to avoid genomic DNA amplification. Total
RNA isolation was performed using RNA TRIzol according to the
manufacturer’s instructions (Invitrogen Life Technologies). By
using the cDNA library construction kit (Clontech, Mountain View,
CA, USA), 1 μg of total RNA was used to synthesize cDNA according
to the manufacturer’s instructions (Takara Bio, Inc., Shiga,
Japan). The transcriptional levels of macroH2A, cyclin D1, cyclin
D3, CDK4, CDK6, CDK8 and GAPDH (a housekeeping gene) were
quantified using the ABI 7500 real-time PCR system (Applied
Biosystems). The amplification conditions were as follows: 95°C for
10 min, followed by 40 cycles of 15 sec at 95°C and 1 min at 60°C,
using the TaqMan® Universal PCR Master mix (Applied
Biosystems). All the results were normalized to the levels of GAPDH
RNA (TaqMan probes; Applied Biosystems). The relative expression
level was calculated using the 2−ΔΔCt method.
Western blot analysis
Samples were separated in 10% SDS-PAGE gels and
transferred to polyvinylidene fluoride membranes (Millipore,
Billerica, MA, USA). Following inhibition with 4% non-fat milk,
macroH2A, cyclin D1, cyclin D3, CDK4, CDK6 and CDK8 were detected
by incubation with the monoclonal anti-macroH2A antibody produced
from rabbit (ab83782; Abcam, Cambridge, MA, USA), anti-cyclin D1
antibody produced from rabbit (ab7958; Abcam), anti-cyclin D3
antibody produced from rabbit (ab112034; Abcam), anti-CDK4 antibody
produced from rabbit (ab7955; Abcam), anti-CDK6 antibody produced
from rabbit (ab151247, Abcam) and anti-CDK8 antibody produced from
rabbit (ab123940; Abcam). GAPDH (a housekeeping gene) was detected
by the monoclonal anti-GAPDH antibody (ab9485, Abcam). The
anti-mouse IgG secondary antibody conjugated to horseradish
peroxidase (Amersham Biosciences, Uppsala, Sweden) and an enhanced
chemiluminescent substrate (ECL plus; Amersham Pharmacia Biotech,
Piscataway, NJ, USA) were used for signal development. Images were
captured using a Fujifilm FLA-5000 image reader (Fujifilm,
Stanford, CT, USA).
Immunofluorescence
B16 melanoma cells were grown on 24×24-mm cover
glasses and then fixed in 4% paraformaldehyde solution in
phosphate-buffered saline (PBS) for 30 min prior to 30 min
incubation with a blocking reagent (5% fetal bovine serum in PBS).
Primary antibodies were incubated with melanoma cells overnight at
4°C prior to washing with PBS. Immunofluorescence staining was
performed with secondary antibodies conjugated to fluorescein
isothiocyanate (F5262; Sigma-Aldrich, St. Louis, MO, USA). A
conventional fluorescence microscope (Carl Zeiss, Göttingen,
Germany) was used for visualization.
Statistical analysis
All values are presented as the mean ± standard
deviation. Continuous variables that did not have a Gaussian
distribution were log transformed. Student’s t-test was used to
compare differences between groups. One-way analysis of variance
was used to determine differences among groups. P≤0.05 was
considered to indicate a statistically significant difference. If F
ratios exceeded the critical value (P≤0.05), the Newman-Keuls post
hoc test was performed to compare the groups.
Results
Overexpression and transfer of macroH2A
in B16 melanoma cells
To detect the effect of macroH2A on B16 melanoma
cells, the vectors of macroH2A interference and overexpression were
designed and then transferred into B16 melanoma cells. qPCR results
demonstrated that the expression levels were the highest in the
macroH2A overexpression group followed by the control group, the
macroH2A overexpression rescue group and the macroH2A interference
rescue group. The lowest expression was identified in the macroH2A
interference group (Fig. 1A).
Differential protein expression was assayed by western blotting.
Similarly, the interference group demonstrated the lowest protein
expression while other groups exhibited a higher expression. In
addition, the overexpression group demonstrated the highest protein
expression among all the groups (Fig.
1B). Immunofluorescence analyses of macroH2A expression in B16
melanoma cells is shown in Fig.
1C. The results also demonstrated the different expression
levels of protein among these treatment groups.
Regulation of B16 melanoma cell line
proliferation by macroH2A
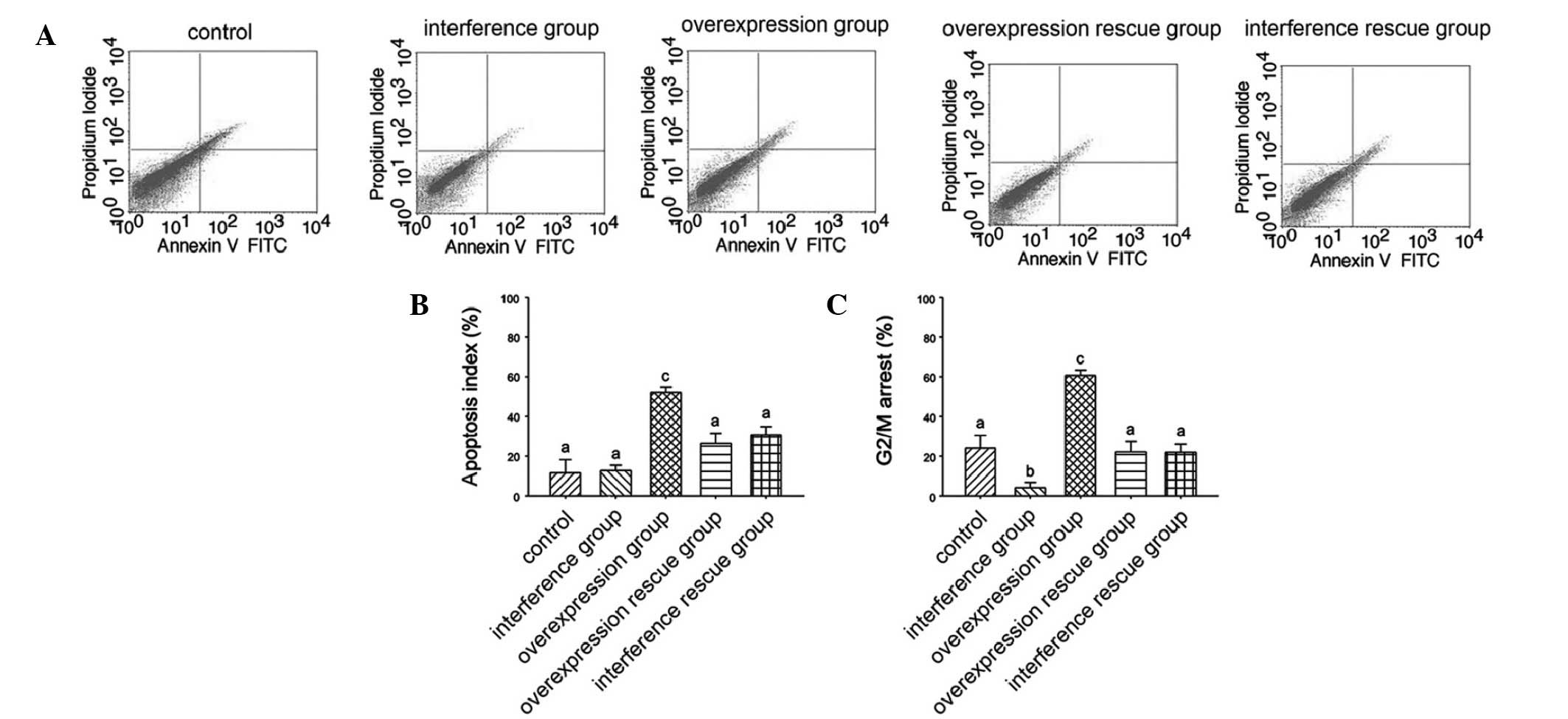
To elucidate the potential cellular regulation of
growth inhibition by macroH2A, cell cycle progress and cell
apoptosis were examined by flow cytometry (Fig. 2A). Apoptosis was highest in the
overexpression group while the apoptosis index was lower in other
groups (Fig. 2B). In addition,
G2/M arrest rate was analyzed. The overexpression group
demonstrated a significantly higher G2/M arrest rate, which
suggested that the overexpression of macroH2A arrested B16 melanoma
cells in the G2/M stage (Fig.
2C).
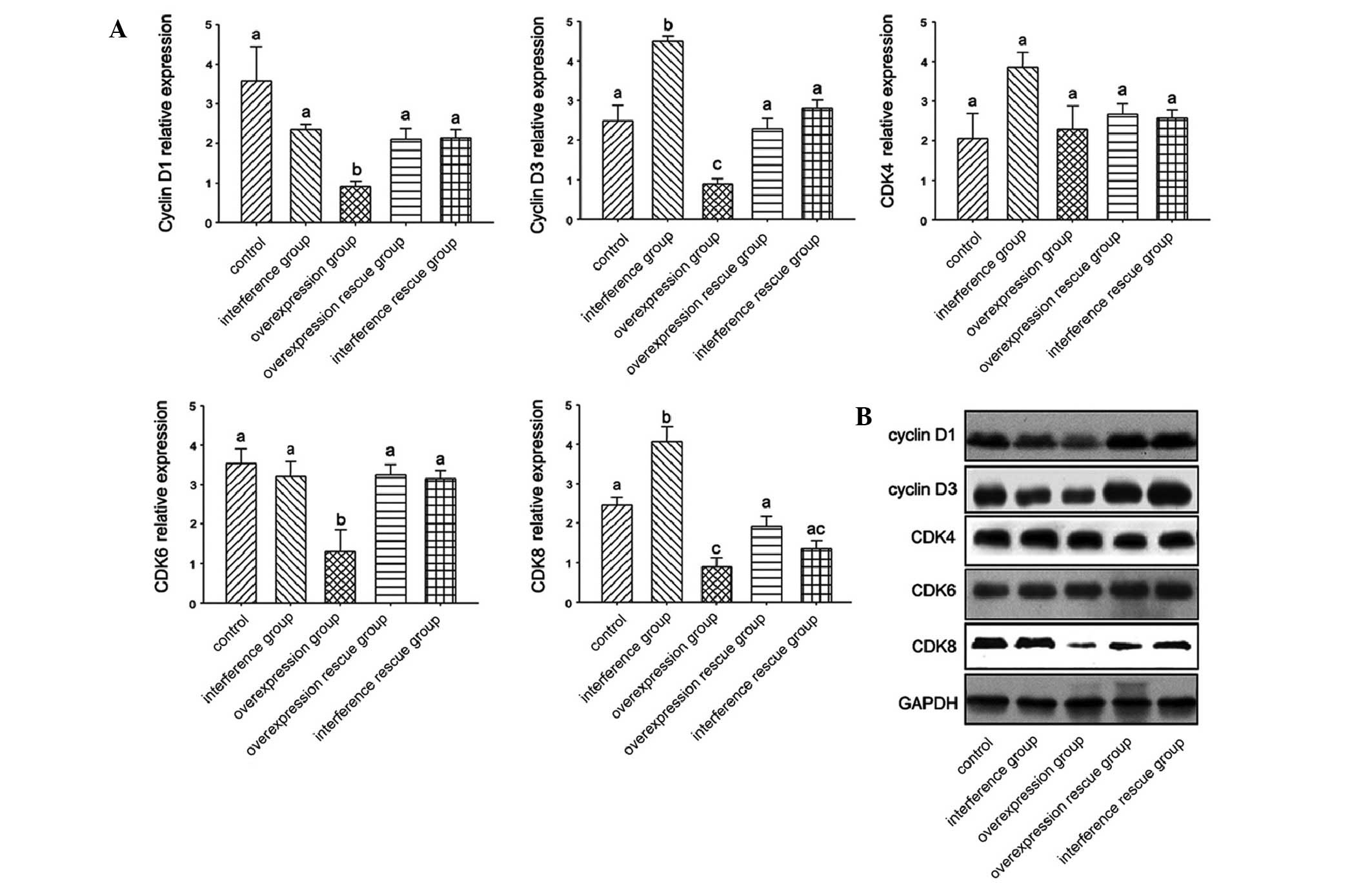
MacroH2A regulates cyclin D and CDKs
To detect the mechanism of inhibition by macroH2A in
B16 melanoma cells, the gene expression of cyclin D and CDKs was
assayed following interference or overexpression of macroH2A.
Cyclin D1 and cyclin D3 mRNA and protein expression levels were
suppressed by the overexpression of macroH2A, while the
interference of macroH2A demonstrated no significant difference in
the expression of cyclin D1 and promoted the expression of cyclin
D3, respectively (Fig. 3A). The
overexpression and interference rescue groups demonstrated no
difference compared with the control group.
CDKs were also analyzed by qPCR and western blot
analysis. No differences among all the treatment groups were
identified in the CDK4 expression. However, among all the treatment
groups, the overexpression group demonstrated a decrease in CDK6.
For CDK8 expression, the interference group demonstrated an
increase in expression, while the overexpression group had a
decreased expression. Similarly, with cyclin D, the overexpression
and interference rescue groups demonstrated no difference compared
with the control group (Fig.
3B).
Discussion
In the present study, the artificial altering of
macroH2A expression was performed by the interference and
overexpression of macroH2A, as well as overexpression rescue and
interference rescue treatments in B16 melanoma cell line
progression, in order to evaluate the impact of altered expression
on melanoma progression and development. It was demonstrated that
macroH2A inhibits the progression of B16 melanoma cells, while
macroH2A knockdown promoted melanoma progression. Similar results
have previously been reported in other forms of human cancer,
including testicular, ovarian, lung, bladder, cervical, breast,
colon and endometrial cancer (25–30).
In these cancer types, the expression levels of macroH2A decreased
significantly in tumor tissues compared with normal tissues. In
addition, in melanoma, macroH2A expression levels demonstrated a
strong negative correlation with tumor development (13). Genome level alterations in macroH2A
also affect cancer progression. Dardenne et al (31) elucidated that the alternative
splicing of macroH2A induced metastasis in breast cancer cell
lines.
The overexpression of macroH2A appears to
participate in the development of malignant tumors, based on its
well-known inhibitory role in cell cycle progression. In addition,
macroH2A also has a role in genomic stabilization during
replication by preventing the occurrence of DNA damage (9). Furthermore, a lack of macroH2A
expression led to an increase in CDK8 expression, which may result
in cell death through avoiding premature mitotic entry (13). The in vitro results of the
present study using overexpression and interference vectors
suggested that the upregulation of macroH2A led to increased cell
death. It is therefore likely that the low expression levels of
macroH2A observed in melanoma tissues prevent DNA damage and cell
death. In accordance with our results, Kapoor et al
(13) previously reported that
macroH2A suppresses melanoma progression through the regulation of
CDK8. In the present study, it was demonstrated that apoptosis of
melanoma cells occurs following the overexpression of macroH2A,
while no significant differences were identified among other
groups. In addition, the G2/M arrest rates were analyzed. The
overexpression group demonstrated a significantly higher G2/M
arrest rate, which suggested the overexpression of
macroH2A-arrested B16 melanoma cells in the G2/M stage. Thus, the
possible explanation of overexpression of macroH2A inducing
apoptosis may be that the cell cycle is disorganized by macroH2A.
The macroH2A expression changes have been demonstrated through S
phase and G2 toward mitosis. This suggested that in this process,
the overexpression of macroH2A arrested melanoma cells in
functional G2/M. In melanoma cells, due to a lack of a functional
G2/M checkpoint caused by the depression of macroH2A, the cell
cycle progresses into mitosis without securing correct DNA
synthesis. Thus, the explanation for the inhibition of the
progression of melanoma by macroH2A may be due to its regulatory
function in the cell cycle.
In the present study, it was revealed that macroH2A
overexpression inhibits cyclin D1, cyclin D3, CDK6 and CDK8 gene
expression. In addition, no significant differences were identified
in CDK4 among all the treatment groups. It has been reported that
cyclin D1 is overexpressed in several cancer types and is regarded
to be an oncogene (32,33). Cyclin D1 affects several mechanisms
in cancer, including the translocation, amplification and
stabilization of mRNA. In addition, elevated cyclin D3 facilitates
cancer progression (34). In the
present study, it was demonstrated that elevated levels of macroH2A
suppressed cyclin D1 and cyclin D3. CDKs, including CDK2, CDK3,
CDK4, CDK5, CDK6, CDK7 and CDK8, are crucial in the cell cycle
(35). Among them, CDK4, CDK6 and
CDK8 are critical for cell proliferation through regulating DNA
synthesis at the beginning of the cell cycle and switching the cell
cycle from G1 to S phase (36).
Since cancer cells often contain high levels of CDK activity,
inhibiting CDK gene expression may be a useful therapeutic strategy
in cancer treatment. It was confirmed that macroH2A depresses CDK6
and CDK8 gene expression in the B16 cell line. Thus, we hypothesize
that other cell cycle-related genes could be regulated by
macroH2A.
In conclusion, the results indicate that the
overexpression of macroH2A induces apoptosis of melanoma cells and
arrests the cell in the G2/M stage of the cell cycle. The data
further demonstrated that, despite being an inhibitor of cell cycle
progression, high expression of macroH2A downregulated cyclin D1,
cyclin D3 and CDK6. Thus, the high expression of macroH2A appears
to protect the cancer cell from a disordered cell cycle through the
regulation of cyclin D1, cyclin D3 and CDK6 genes. The present
findings support emerging links between chromatin structure and
cancer and to the best of our knowledge are the first to
demonstrate a direct role of macroH2A in this process.
Acknowledgements
This study was supported by the Natural Science
Foundation of Hunan Provincial (grant no. 13JJ2011) and the
Scientific Research Fund of Hunan Provincial Health Department
(grant no. hn772705).
References
|
1
|
Portela A and Esteller M: Epigenetic
modifications and human disease. Nat Biotechnol. 28:1057–1068.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Baylin SB and Jones PA: A decade of
exploring the cancer epigenome-biological and translational
implications. Nat Rev Cancer. 11:726–734. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kulaeva OI, Gaykalova DA and Studitsky VM:
Transcription through chromatin by RNA polymerase II: histone
displacement and exchange. Mutat Res. 618:116–129. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Neely KE and Workman JL: The complexity of
chromatin remodeling and its links to cancer. Biochim Biophys Acta.
1603:19–29. 2002.PubMed/NCBI
|
|
5
|
Hake SB, Xiao A and Allis CD: Linking the
epigenetic ‘language’ of covalent histone modifications to cancer.
Br J Cancer. 90:761–769. 2004.
|
|
6
|
Strahl BD and Allis CD: The language of
covalent histone modifications. Nature. 403:41–45. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Peterson CL and Laniel MA: Histones and
histone modifications. Curr Biol. 14:R546–R551. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Redon C, Pilch D, Rogakou E, et al:
Histone H2a variants H2AX and H2AZ. Curr Opin Genet Dev.
12:162–169. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pehrson JR and Fried VA: macroH2A, a core
histone containing a large nonhistone region. Science.
257:1398–1400. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kustatscher G, Hothorn M, Pugieux C, et
al: Splicing regulates NAD metabolite binding to histone macroH2A.
Nat Struct Mol Biol. 12:624–625. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Brooks WH: X chromosome inactivation and
autoimmunity. Clin Rev Allergy Immunol. 39:20–29. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chadwick BP and Willard HF: Histone H2A
variants and the inactive X chromosome: identification of a second
macroH2A variant. Hum Mol Genet. 10:1101–1113. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kapoor A, Goldberg MS, Cumberland LK, et
al: The histone variant macroH2A suppresses melanoma progression
through regulation of CDK8. Nature. 468:1105–1109. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rickert P, Seghezzi W, Shanahan F, et al:
Cyclin C/CDK8 is a novel CTD kinase associated with RNA polymerase
II. Oncogene. 12:2631–2640. 1996.PubMed/NCBI
|
|
15
|
Donner AJ, Szostek S, Hoover JM, et al:
CDK8 is a stimulus-specific positive coregulator of p53 target
genes. Mol Cell. 27:121–133. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sánchez I and Dynlacht BD: New insights
into cyclins, CDKs, and cell cycle control. Semin Cell Dev Biol.
311–321. 2005.PubMed/NCBI
|
|
17
|
Satyanarayana A and Kaldis P: Mammalian
cell-cycle regulation: several Cdks, numerous cyclins and diverse
compensatory mechanisms. Oncogene. 28:2925–2939. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fisher RP: CDKs and cyclins in
transition(s). Curr Opin Genet Dev. 7:32–38. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kastan MB and Bartek J: Cell-cycle
checkpoints and cancer. Nature. 432:316–323. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Andreassi MG: DNA damage, vascular
senescence and atherosclerosis. J Mol Med (Berl). 86:1033–1043.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Morgan DO: Cyclin-dependent kinases:
engines, clocks, and microprocessors. Annu Rev Cell Dev Biol.
13:261–291. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tyson JJ, Csikasz-Nagy A and Novak B: The
dynamics of cell cycle regulation. Bioessays. 24:1095–1109. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sherr CJ and Roberts JM: CDK inhibitors:
positive and negative regulators of G1-phase progression. Genes
Dev. 13:1501–1512. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Taylor WR and Stark GR: Regulation of the
G2/M transition by p53. Oncogene. 20:1803–1815. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sporn JC, Kustatscher G, Hothorn T, et al:
Histone macroH2A isoforms predict the risk of lung cancer
recurrence. Oncogene. 28:3423–3428. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Novikov L, Park JW, Chen H, et al:
QKI-mediated alternative splicing of the histone variant macroH2A1
regulates cancer cell proliferation. Mol Cell Biol. 31:4244–4255.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Haugstetter A, Loddenkemper C, Lenze D, et
al: Cellular senescence predicts treatment outcome in metastasised
colorectal cancer. Br J Cancer. 103:505–509. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Buschbeck M and Di Croce L: Approaching
the molecular and physiological function of macroH2A variants.
Epigenetics. 5:118–123. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang R and Adams PD: Heterochromatin and
its relationship to cell senescence and cancer therapy. Cell Cycle.
6:784–789. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Collado M, Gil J, Efeyan A, et al: Tumour
biology: senescence in premalignant tumours. Nature. 436:6422005.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dardenne E, Pierredon S, Driouch K, et al:
Splicing switch of an epigenetic regulator by RNA helicases
promotes tumor-cell invasiveness. Nat Struct Mol Biol.
19:1139–1146. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shtutman M, Zhurinsky J, Simcha I, et al:
The cyclin D1 gene is a target of the β-catenin/LEF-1 pathway. Proc
Natl Acad Sci USA. 96:5522–5527. 1999.
|
|
33
|
Fu M, Wang C, Li Z, et al: Minireview:
Cyclin D1: normal and abnormal functions. Endocrinology.
145:5439–5447. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hunter T and Pines J: Cyclins and cancer.
II: Cyclin D and CDK inhibitors come of age. Cell. 79:573–582.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gray N, Détivaud L, Doerig C, et al:
ATP-site directed inhibitors of cyclin-dependent kinases. Curr Med
Chem. 6:859–875. 1999.PubMed/NCBI
|
|
36
|
Lapenna S and Giordano A: Cell cycle
kinases as therapeutic targets for cancer. Nat Rev Drug Discov.
8:547–566. 2009. View
Article : Google Scholar : PubMed/NCBI
|