Introduction
The biological repair of articular cartilage defects
is a focus of the study of orthopedics and tissue engineering is
generally considered to be the most promising and effective
approach (1,2). The isolation and cell culture of
articular chondrocytes in vitro aims to obtain a sufficient
amount of the desired seed cells for cartilage tissue engineering
(2). Furthermore, the application
of cytokines aims to accelerate cell proliferation and maintain the
normal function of articular cartilage cells (2). Therefore, investigation concerning
the mechanisms of action of cytokines in cultured chondrocytes
in vitro has significant application potential.
Platelet-derived growth factor (PDGF), a 30 kDa
dimer of A and B polypeptide chains linked by disulphide bonds, is
a multiple mitogen for the regulation of the replication of
osteoblasts and degradation of bone collagens (3,4). A
study by Brandl et al (2)
demonstrated that there was a markedly higher proliferation rate of
chondrocytes cultured with PDGF-BB and transforming growth factor
(TGF)-β1 than that of the control cells. Schmidt et al
(4) revealed that PDGF was able to
promote chondrocyte proliferation and proteoglycan synthesis. It
has also been reported that PDGF is able to stimulate cultured
chondrocytes and repair cartilage tissue proliferation (5,6).
PDGF was demonstrated to be the autocrine cytokine of human
osteoblasts (3). Therefore, the
present study focused on its effects on chondrocyte formation.
To date, few studies have investigated the specific
mechanisms underlying the action of PDGF in cartilage cells. Rui
et al (7) revealed that the
overexpression of G-protein-coupled receptor kinase interacting
protein-1 (GIT1) induced by PDGF was able to activate the
extracellular signal-regulated kinase (ERK) 1/2 pathway to promote
osteoblast migration and bone healing. GIT1, a multidomain scaffold
protein, is involved in the internalization and membrane
transportation of the G protein-coupled receptor (8). Pang et al (8) demonstrated that the interaction
between mitogen-activated protein kinase (MAPK)-1 and GIT1 was able
to activate ERK1/2. Additionally, the translocation of ERK1/2 and
activation in focal adhesions are responsible for cell spreading
and migration with the binding of GIT1 (7,8).
Certain studies have demonstrated that GIT1 is expressed in
osteoblasts and osteoclasts, implying that GIT1 may be involved in
bone metabolism (7,9,10).
Additionally, even though there were no significant effects on the
differentiation and function of mouse osteoblasts in the absence of
GIT1, the GIT1 Y321F mutant was able to inhibit PDGF-induced
osteoblast movement and migration, according to the Boyden chamber
assay (7,10). The studies suggested that the
phosphorylation of tyrosine 321 of GIT1 is closely associated with
the combination of focal adhesion kinase and the migration of
osteoblasts induced by PDGF (7,10).
The formation of pseudopodia bodies in osteoclasts without GIT1
resulted in abnormal bone resorption and destruction capabilities
and affected the regulation of the osteoclast cytoskeleton receptor
activator of nuclear factor-κB.
In addition, GIT1, apart from interacting with ERK-1
and activating ERK1/2, is also essential for the phosphorylation of
phospholipase Cγ1 (PLCγ1) (8,11).
Furthermore, Ren et al (11) revealed that cyclic mechanical
stress may promote PLCγ1 phosphorylation and activate the ERK1/2
signaling pathway for rat chondrocyte proliferation and
extracellular matrix synthesis. PLCγ1, one of the serine threonine
kinases of PLC, is important in the regulation of various cellular
responses generated by stimuli in the signal transduction pathway
(12). Husain et al
(13) demonstrated that the
proliferative activity of endothelial cells during overexpression
of vascular endothelial growth factor receptor-2 depended on the
activation of PLCγ1. Hunter et al (14) demonstrated that PDGF activated the
PLCγ1-mediated ERK1/2 signaling pathway and ultimately regulated
the transcription of vascular smooth muscle cells (14). Thus, in the present study, it was
hypothesized that PDGF is able to activate the ERK1/2 pathway for
the regulation of chondrocyte proliferation and apoptosis through
the promotion of GIT1 expression and PLCγ1 phosphorylation. As a
result, therefore, the aim of the present study was to verify the
above hypothesis at the cellular level, based on the isolation and
cell culture of chondrocytes.
Materials and methods
Materials
Sprague-Dawley neonatal rats were purchased from the
Institute of Laboratory Animal Sciences, Chinese Academy of Medical
Sciences and Peking Union Medical College (Beijing, China). The
present study was approved by the ethics committee of Liuhuaqiao
Hospital (Guangzhou, China). Dulbecco’s modified Eagle’s medium
(DMEM) was obtained from Invitrogen Life Technologies (Carlsbad,
CA, USA). The MEK1/2 inhibitor PD98059 was purchased from Cell
Signaling Technology (Beverly, MA, USA) and the PLCγ1 inhibitor
U73122 was obtained from Santa Cruz Biotechnology, Inc. (Santa
Cruz, CA, USA). The whole antibodies used in the present study were
purchased from Cell Signaling Technology and thiazolyl blue
tetrazolium bromide and propidium iodide (PI) were purchased from
Sigma-Aldrich (St. Louis, MO, USA).
Cell culture
One-week-old neonatal Sprague-Dawley rats were
dissected and their limb joints were extracted under sterile
conditions. The fibrous tissue of the articular cartilage surface
was stripped carefully and the cartilage was cut from the
transparent central portion into pieces of ~1 mm3 in
size. The tissue pieces were incubated with trypsin (0.25%) at 37°C
for 30 min. The supernatant was discarded following centrifugation
at 10,000 × g for 5 min. Centrifugation using a 200 mesh filter and
at 10,000 × g for 5 min was performed to obtain the pellet.
Following that, 0.2% collagenase II was added for digestion and
incubated at 37°C for 4 h. Finally, the cells were maintained in
DMEM containing FBS (10%), penicillin (100 U/ml) and streptomycin
(50 U/ml) at 37°C in a humidified incubator with 5% CO2,
respectively. The morphology of the cells purified by repeated
isolation and culture was observed under an inverted phase contrast
microscope. Once the cells had grown completely in the dish, 0.25%
trypsin was used for digestion and passage. Then, 105
cultured cartilage cells were collected and seeded in a six-well
plate. The cells were rinsed with phosphate-buffered saline (PBS)
when they were adherent. Following fixing with 4% paraformaldehyde
at 4°C for 30 min, the conventional avidin-biotin complex method
for type II collagen staining was performed and the stained cells
were observed under an Olympus IX-70 microscope (Olympus, Tokyo,
Japan).
Transfection of small interfering (si)
RNA
The cells were seeded at a density of
1×105 cells/ml into a six-well plate and incubated for
24 h. siRNA transfection was performed according to the
manufacturer’s instructions of Lipofectamine 2000 (Invitrogen Life
Technologies), when the cell confluence reached ~70%. The siRNA
sequences used were: GIT1 forward, 5′-AAGCTGCCAAGAAGAAGCTAC-3′ and
reverse, 5′-AATTCTCCGACACGTGTCACT-3′. GIT1 siRNA was prepared and
transfected at 100 nM for 24 h as previously described (4,5). The
MEK1/2 inhibitor PD98059 and PLCγ1 inhibitor U73122 were dissolved
in dimethyl sulfoxide (DMSO; Sigma-Aldrich). After the cells were
pretreated with PD98059 (30 nM), U73122 (1 μM) or the same volume
of 0.1% DMSO for 1 h, 10 ng/ml of PDGF (Sigma-Aldrich) was added to
stimulate for 24 h. Following 72 h, transfected cells were analyzed
by quantitative polymerase chain reaction (qPCR) and western blot
analysis.
Western blot analysis
The lysis buffer was used for the extraction of
total cellular protein from the cells. Following adding lysis
buffer, the mixture was agitated at 4°C for 20 min. The protein in
the cell lysates was collected from the supernatant following
centrifugation at 10,000 × g at 4°C for 10 min. The protein
concentrations in the cell lysates were determined by the Bradford
protein assay. SDS-PAGE was conducted in 8% glycine gels (Bio-Rad,
Hercules, CA, USA) loading equal amounts of proteins per lane.
Following electrophoresis, the nitrocellulose membrane was used for
transformation to separate proteins. Then, the nitrocellulose
membrane was inhibited with 5% non-fat milk in Tween 20 with
Tris-buffered saline (TBST) buffer for 1 h. Following that, the
membranes were incubated with primary antibodies at a dilution of
1:1,000 overnight at 4°C and then anti-rabbit immunoglobulin G
monoclonal antibody conjugated with horseradish peroxidase (Cell
Signaling Technology) at a dilution of 1:7,000–8,000 was incubated
for 1 h at 37°C. TBST was used for washing the membranes every 10
min, for a total of 30 min. The protein bands were detected using
enhanced chemoluminescence. The light-emitting films were scanned
by a GelBlot-Pro 1.01 gel imaging system (UVP, Inc., Upland, CA,
USA) for western blotting and gray values of the bands were
measured by Gel-Pro Analyzer software 6.3 (Media Cybernetics,
Rockville, MD, USA).
Bromodeoxyuridine (BrdU) cell
proliferation assay
To examine the roles of PDGF, PLCγ1, GIT1 and the
ERK1/2 pathway on cell proliferation, the effects of different
inhibitors on cell proliferation were examined by a BrdU assay.
Briefly, the cells were seeded into a six-well plate and incubated
for 24 h, and then siRNA transfection was performed using
Lipofectamine 2000. Following pretreatment of the cells with 50 μM
NSC23766, 30 nM PD98059 or the same volume of 0.1% DMSO for 1 h, 10
ng/ml PDGF was then added to stimulate the cells for 24 h. A BrdU
colorimetric immunoassay kit (Cell Proliferation ELISA; Roche
Diagnostics, Mannheim, Germany) was used for the quantification of
cell proliferation according to the manufacturer’s instructions.
The cell proliferation was expressed as the mean percentage of the
control values (set at 100%).
Flow cytometric analysis
The cells were trypsinized and washed with PBS.
Following that, the cells were fixed with 75% ethanol overnight at
−20°C. The next day, PBS was used to wash the fixed cells.
Following that, propidium iodide (PI) working solution (1.21 mg/ml
Tris; 700 U/ml RNase; 50.1 μg/ml PI; pH 8.0) was stained with the
fixed cells for 4 h in the dark. An Epics XL-MCL flow cytometer
(Beckman Coulter, Miami, FL, USA) was used to analyze the stained
cells. The cell cycle distribution was analyzed using MultiCycle
software 5.0 (Phoenix Flow Systems, San Diego, CA, USA). The
proportion of cells in G0/G1, S and G2/M phases are presented as
DNA histograms. The apoptotic cells with hypodiploid DNA contents
were measured by quantifying the sub-G1 peak in the cell cycle
pattern. For each experiment, 10,000 events per sample were
recorded.
Terminal deoxynucleotidyl transferase
dUTP nick end labeling (TUNEL)-4′,6-diamidino-2-phenylindole (DAPI)
co-staining assay
Apoptotic cells were assessed by a TUNEL-DAPI
co-staining assay using an In Situ Cell Death Detection kit (Roche
Diagnostics) according to the manufacturer’s instructions. Briefly,
cultured cells were washed with PBS and then fixed with 4%
formaldehyde at 4°C for 25 min. The cells were then incubated with
0.2% Triton X-100 for 5 min. Following incubation with 100 μl of
equilibration buffer at room temperature for 10 min, the cells were
mixed with 50 μl of TUNEL reaction mixture containing nucleotide
mixture and terminal deoxynucleotidyl transferase for 60 min at
37°C. The cells were washed with 2× saline sodium citrate for 15
min. The cells were then incubated with 0.3%
H2O2 for 10 min and with streptavidin working
solution for 30 min at room temperature, respectively. Then, 0.5
μg/ml of DAPI was added and cells were incubated in a humidified
chamber in the dark for 5 min at room temperature. Finally, they
were examined and images were captured under a fluorescence
microscope (Nikon Eclipse 80i; Nikon, Tokyo, Japan).
Statistical analysis
The experiments were performed at least in
triplicate and the results are expressed as the mean ± standard
deviation. SPSS statistical package (SPSS 13.0 for Windows; SPSS,
Inc., Chicago, IL, USA) was used for statistical analysis. The
difference between two groups was analyzed by a two-tailed
Student’s t-test and that between three or more groups was analyzed
by one-way analysis of variance multiple comparisons.
*P<0.05 or **P<0.01 was considered to
indicate a statistically significant difference.
Results
PDGF increases the expression of GIT1 to
promote the phosphorylation of PLCγ1 and ERK
In the present study, the effects of PDGF, GIT1,
PLCγ1, ERK1/2 and their crosstalk in chondrocytes were investigated
by siRNA silencing techniques and western blot analysis. The cells
were seeded and cultured in a six-well plate for 24 h, followed by
siRNA transfection of GIT1 siRNA using Lipofectamine 2000.
Following pretreatments of the cells with siRNA transfection or
NSC23766, PD98059 and DMSO for 1 h, 10 ng/ml of PDGF was then added
to stimulate the cells for 24 h. The expression levels of
associated proteins were examined by western blot analysis. As
shown in Fig. 1, the results
demonstrated that PDGF was able to increase the expression of GIT1
by 2.8-fold, thereby promoting the phosphorylation of PLCγ1 and
activation of the ERK1/2 pathway by 3.0 and 4.0-fold, respectively.
In the group in which GIT1 was depleted by siRNA, even though PDGF
was added to the chondrocytes for stimulation, the expression
levels of GIT1 decreased to 60% compared with those in the control.
In addition, the PLCγ1 inhibitor U73122 or the ERK1/2 pathway
inhibitor PD98059 did not affect GIT1 expression significantly,
suggesting that GIT1 was upstream of PLCγ1. Furthermore, PDGF
stimulation was able to induce the overexpression of PLCγ1 and the
phosphorylation of ERK1/2. However, PDGF did not markedly increase
the expression of PLCγ1 and the phosphorylation of ERK1/2 when GIT1
expression was inhibited. The results demonstrated that PDGF was
not able to stimulate the upregulation of ERK1/2 due to the
suppression of PLCγ1. Therefore, PDGF first upregulated the
expression of GIT1, then activated PLCγ and eventually activated
the ERK1/2 pathway.
Roles of PLCγ1, GIT1 and ERK1/2 in
PDGF-activated cell proliferation
To further examine the biological functional roles
of PLCγ1, GIT1 and ERK1/2 in chondrocytes, the effects of
inhibition of PDGF-activated cell proliferation by RNA interference
or small molecule inhibitors were investigated. Briefly, the cells
were seeded into a six-well plate and incubated for 24 h, and then
GIT1 siRNA transfection was performed using Lipofectamine 2000.
Following pretreatment of the cells with siRNA transfection, 50 μM
NSC23766, 30 nM PD98059 or the same volume of 0.1% DMSO for 1 h, 10
ng/ml PDGF was then added to stimulate the cells for 24 h, and then
the cell proliferation was examined by a BrdU assay. As shown in
Fig. 2, PDGF was able to promote
the cell proliferation of chondrocytes through the GIT1- and
PLCγ-mediated ERK1/2 pathway and GIT1 may be essential in the
activation of PLCγ and ERK1/2. The investigation of the effects of
the ERK1/2 inhibitor PD98059 and PLCγ inhibitor U73122 on
chondrocyte proliferation verified that chondrocyte proliferation
was significantly decreased by inhibition of either PLCγ or ERK1/2.
Based on these results, it is possible that there are certain other
pathways involved in the induction of chondrocyte proliferation by
PDGF. At present, there are no studies comparing the effects of
inhibiting the phosphorylation of PLCγ and the inhibition of the
expression of ERK1/2. Chondrocyte proliferation was increased by
PDGF when PLCγ phosphorylation or ERK1/2 expression were inhibited,
indicating that additional proteins may be involved in PDGF-induced
growth stimulation.
 | Figure 2Roles of PLCγ1, GIT1 and ERK1/2 in
PDGF-activated cell proliferation. Following pretreatment of the
cells with siRNA transfection, 50 μM NSC23766, 30 nM PD98059 or the
same volume of 0.1% DMSO for 1 h, 10 ng/ml PDGF was then added to
stimulate for 24 h. The cell proliferation was examined by BrdU
assay. 1, Control; 2, PDGF; 3, PDGF + NC siRNA; 4, PDGF + GIT1
siRNA; 5, PDGF + U73122; 6, PDGF + PD98095; 7, PDGF + DMSO. PDGF,
platelet-derived growth factor; GIT1, G-protein-coupled receptor
kinase interacting protein-1; PLCγ1, phospholipase Cγ1; ERK,
extracellular signal-regulated kinase; DMSO, dimethyl sulfoxide;
siRNA, small interfering RNA; NC, negative control; BrdU,
bromodeoxyuridine. a, b, c and d indicate a statistically
significant difference between each other (P<0.01). |
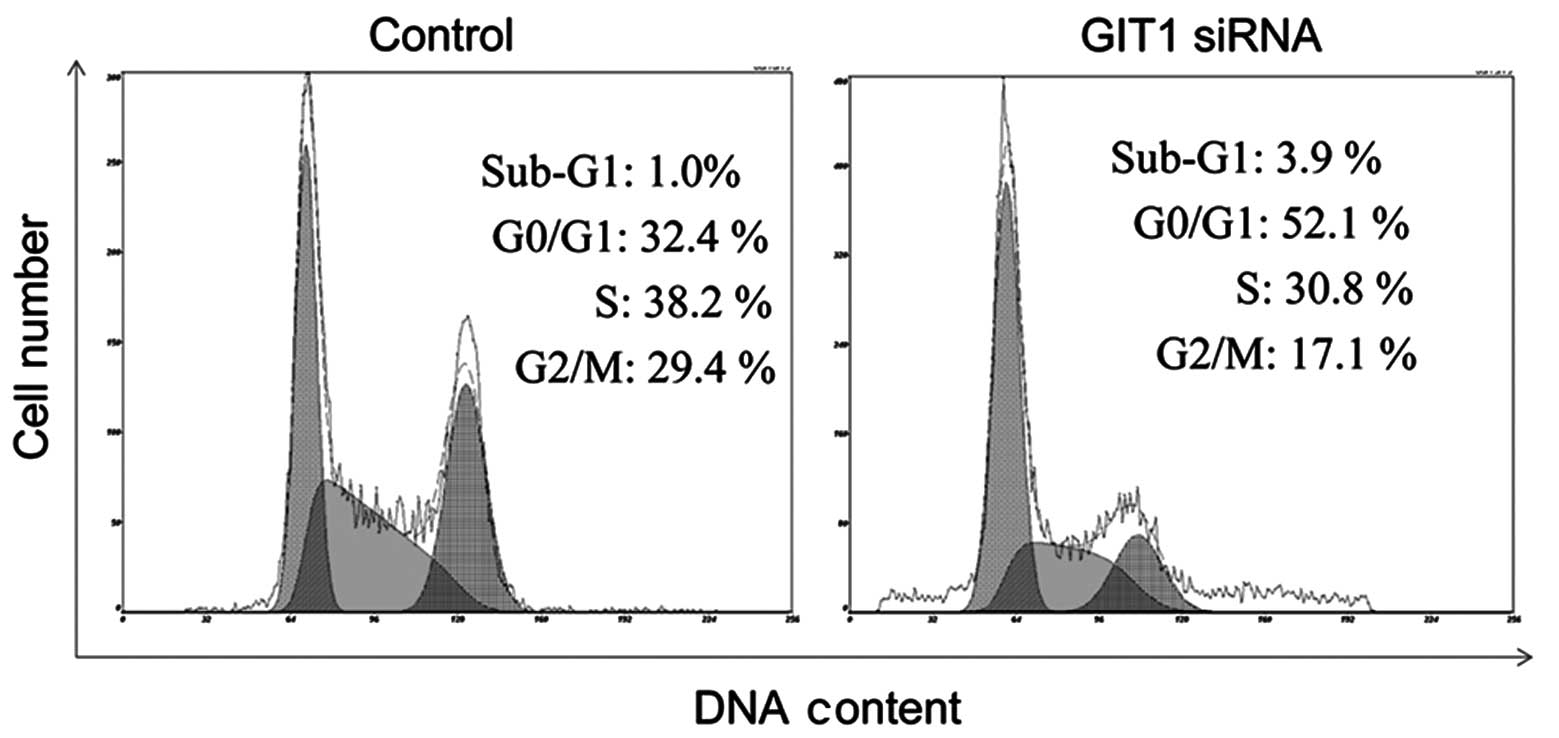
PDGF affects the cell cycle of
chondrocytes
The effects of GIT1 knockdown on cell cycle
distribution were analyzed by flow cytometry. Cell cycle analysis
by quantitation of DNA content was one of the earliest applications
of flow cytometry. The DNA of mammalian cells is able to be stained
by a variety of DNA binding dyes, including PI. The premise with
these dyes is that they are stoichiometric i.e. they bind in
proportion to the amount of DNA present in the cell. In this way
cells that are in the S phase have more DNA than cells in G1 and,
therefore, they take up proportionally more dye and fluoresce more
brightly until they have doubled their DNA content. Cells in G2
phase incorporate approximately twice as much stain as cells in G1
phase. As shown in Fig. 3,
following depletion of GIT1, the number of cells in G0/G1 phase
increased significantly from 32.4 (control) to 52.1%, with a slight
decrease of cells in S phase. This inhibition of cell cycle
progression explains the observed decreases in cell
proliferation.
Effect of PLCγ1, GIT1 and ERK1/2 on
apoptosis
Apoptosis is the process of programmed cell death
that occurs in multicellular organisms. It has been verified that
apoptosis in chondrocytes is important for bone remodeling
(15). The TUNEL-DAPI assay is a
common method for detecting DNA fragmentation that results from
apoptotic signaling cascades. The assay relies on the presence of
nicks in the DNA which are able to be identified by the TUNEL
assay, which was used to elucidate the role of GIT1 in chondrocyte
apoptosis in the present study. The results (Fig. 4) demonstrated that apoptosis, as
evidenced by DNA fragmentation, was observed in chondrocytes.
Notably, the addition of PDGF was able to effectively inhibit the
occurrence of apoptotic cell death. Furthermore, depletion of GIT1
and PLCγ1 effectively reversed the protective effects of PDGF.
However, the inhibition of ERK1/2 demonstrated no significant
effect on apoptosis. Taken together, these results suggested that
GIT1 and PLCγ1 are necessary for the survival of chondrocytes
induced by PDGF, while the ERK1/2 pathway was not important in the
anti-apoptotic effect of PDGF on chondrocytes.
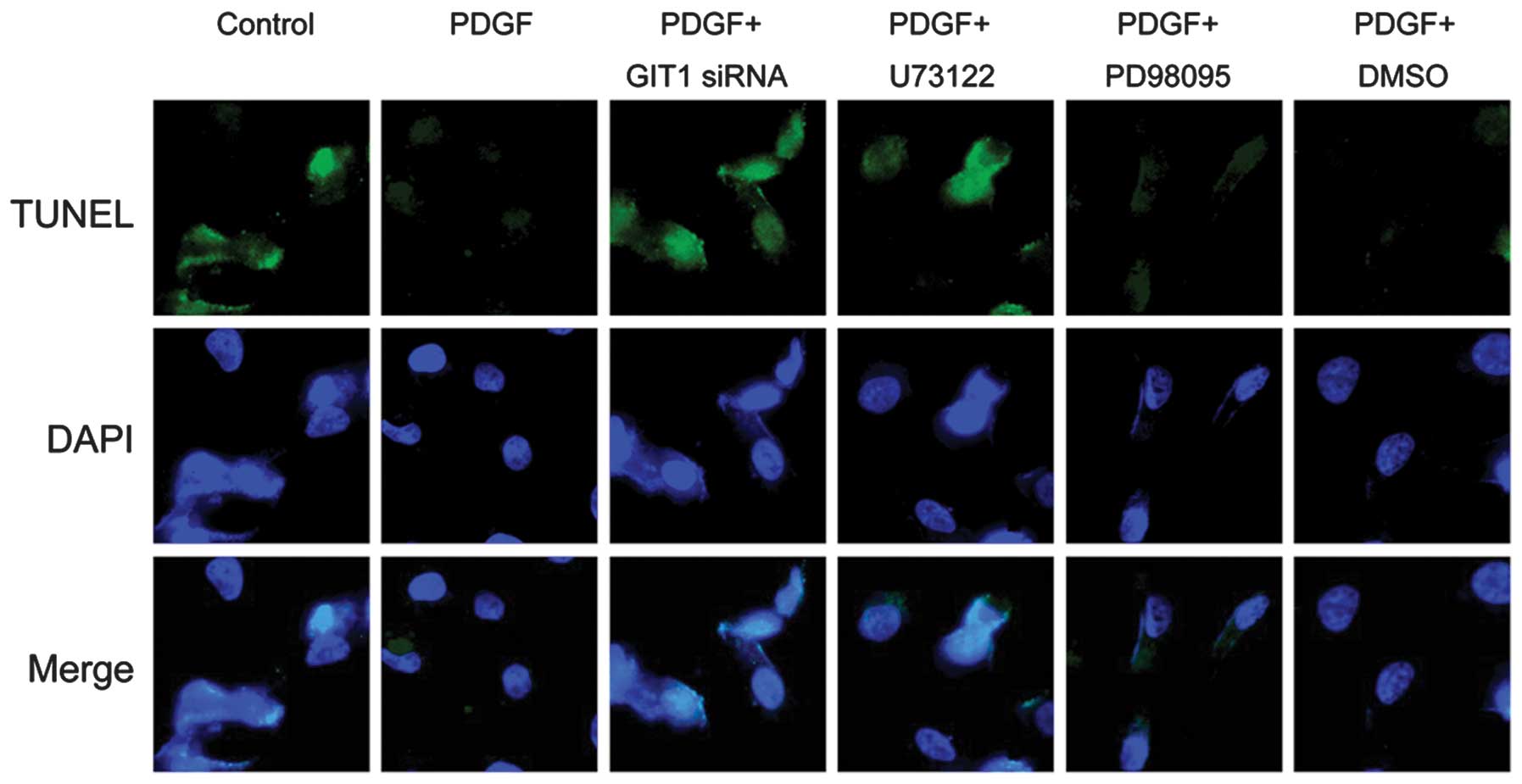 | Figure 4Effects of GIT1, PLCγ1 and ERK1/2 on
the anti-apoptotic activity of PDGF in chondrocytes. Following
pretreatment of the cells with siRNA transfection and various
specific inhibitors, 10 ng/ml PDGF was then added to stimulate for
24 h. Apoptosis was determined by a TUNEL-DAPI co-staining assay.
PDGF, platelet-derived growth factor; GIT1, G-protein-coupled
receptor kinase interacting protein-1; PLCγ1, phospholipase Cγ1;
ERK, extracellular signal-regulated kinase; TUNEL, terminal
deoxynucleotidyl transferase dUTP nick end labeling; DAPI,
4′,6-diamidino-2-phenylindole; DMSO, dimethyl sulfoxide; siRNA,
small interfering RNA. |
Discussion
PDGF, one of the growth factors involved in the
early stage of healing of damaged tissue, is an essential growth
factor for the healing process (16,17).
PDGF is able to stimulate cartilage DNA and protein synthesis
(18). PDGF is also known to
increase the IGF1 production of mesenchymal cells and was able to
stimulate chondrocyte proteoglycan synthesis by IGF1 (19). At present, there have been few
studies investigating the complete mechanisms of PDGF in cartilage
cells.
According to the studies of Rui et al
(7) and Ren et al (11), the present study hypothesized that
PDGF is able to activate the ERK1/2 pathway for the regulation of
chondrocyte proliferation and apoptosis through the promotion of
GIT1 expression and PLCγ1 phosphorylation. In the present study,
PDGF was used to stimulate the cultured chondrocytes in
vitro to investigate its effects. The results indicated that
GIT1 was overexpressed and PLCγ1 and ERK1/2 were induced to undergo
phosphorylation. It was revealed that the induction of PLCγ1 and
ERK1/2 phosphorylation was not significant when PDGF was knocked
down by siRNA. In other words, GIT1 was involved in the ERK1/2
pathway activated by PDGF and was able to activate PLCγ1 and ERK1/2
phosphorylation. Following the application of the PLCγ1 inhibitor
U73122 and the ERK1/2 inhibitor PD98059, the expression of GIT1 did
not alter significantly, suggesting that GIT1 is upstream of PLCγ1.
In addition, activation of the ERK1/2 signaling pathway induced by
PDGF was not significant when PLCγ1 was inhibited by U73122. Thus,
activation of the ERK1/2 signaling pathway by PDGF proceeds via
upregulated expression of PDGF, activation of GIT1 followed by
activation of PLCγ1, and eventually the activation of the ERK1/2
pathway. This is consistent with the hypothesis of the present
study.
Based on the results on the cell proliferation of
chondrocytes, PDGF stimulated the proliferation of chondrocytes,
which was markedly attenuated following the downregulation of GIT1.
The ability of PDGF to promote proliferation was also inhibited
when PLCγ1 phosphorylation and the ERK1/2 pathway were blocked.
However, the inhibitory effect on cell proliferation by inhibition
of the ERK1/2 pathway was lower than the effect of inhibition of
GIT1 and PLCγ1. These results revealed that GIT1 and PLCγ1 may
promote chondrocyte proliferation through other pathways. The study
by Ren et al (11) verified
that GIT1, as a scaffold protein, is closely associated with PLCγ1
and the ERK1/2 pathway. Furthermore, GIT1 was able to control the
regulation of chondrocyte proliferation, migration and apoptosis
through the PLCγ1 and ERK1/2 pathway (20). Based on studies on osteoblasts,
PDGF is vital in osteoblast proliferation and differentiation and
is able to promote osteoblast DNA synthesis at an early stage,
leading to the transition of osteoblasts from stationary G0/G1 into
the replicative S phase. It also enhances the migration of
monocytes and fibroblasts, so that local fibroblast proliferation
and differentiation can be induced to promote bone formation. Yang
et al (7) demonstrated that
the number of osteoblasts in G0/G1 phase was reduced, while that in
the S phase was increased in human fetal skull cells in
vitro. In addition, PDGF-AA, PDGF-BB and TGF were able to
enhance the expression of PDGF-A mRNA (7). This demonstrated that PDGF-AA is able
to accelerate the cell cycle and induce the division of quiescent
cells to enhance cell replication, thereby promoting bone
formation. Based on the results of a space study by Kumei et
al (20), PDGF-β, epidermal
growth factor and its receptor binding are able to be adjusted by
the microgravity in space caused by decreased osteoblast function,
and are able to activate tyrosine kinases to bind with the adapter
protein Shc Grb2. The induction of Ras/MAPK is able to stimulate
signaling through different chemical and physical reactions to the
nucleus, leading to the induction of c-fos and c-jun gene
expression (20).
The present study demonstrated that PDGF stimulation
reduced chondrocyte apoptosis. However, downregulation of GIT1
expression and inhibition of the phosphorylation of PLCγ1 is able
to inhibit the effects of PDGF. By contrast, inhibition of the
ERK1/2 pathway did not significantly affect chondrocyte apoptosis.
Previous studies have identified GIT1 as a regulator of
mitochondrial biogenesis and function, which is required for
postnatal cardiac maturation (21). The present study also demonstrated
that GIT1 is involved in the proliferation of chondrocytes, which
is an important process for fracture healing. Furthermore, a study
by Zhang et al (22)
indicated that GIT1 inhibited apoptosis via the modulation of the
inositol triphosphate receptor-mediated Ca2+ signal
(22). Therefore, the suppression
of chondrocyte apoptosis by PDGF was mediated by the promotion of
the expression of GIT1 and the phosphorylation of PLCγ1; however,
it did not proceed via the ERK1/2 pathway.
In conclusion, PDGF was able to promote chondrocyte
proliferation and inhibit apoptosis through the promotion of the
expression of GIT1 and the phosphorylation of PLCγ1. The
stimulation of proliferation by PDGF was achieved through the
ERK1/2 pathway; however, the results indicated that other pathways
were also involved. By contrast, the suppression of apoptosis by
PDGF did not proceed via the ERK1/2 pathway.
References
|
1
|
Filardo G, Kon E, Di Martino A, Iacono F
and Marcacci M: Arthroscopic second-generation autologous
chondrocyte implantation: a prospective 7-year follow-up study. Am
J Sports Med. 39:2153–2160. 2011.
|
|
2
|
Brandl A, Angele P, Roll C, Prantl L,
Kujat R and Kinner B: Influence of the growth factors PDGF-BB,
TGF-beta1 and bFGF on the replicative aging of human articular
chondrocytes during in vitro expansion. J Orthop Res. 28:354–360.
2010.
|
|
3
|
Yang D, Chen J, Jing Z and Jin D:
Platelet-derived growth factor (PDGF)-AA: a self-imposed cytokine
in the proliferation of human fetal osteoblasts. Cytokine.
12:1271–1274. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schmidt MB, Chen EH and Lynch SE: A review
of the effects of insulin-like growth factor and platelet derived
growth factor on in vivo cartilage healing and repair.
Osteoarthritis Cartilage. 14:403–412. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Barbero A, Palumberi V, Wagner B, Sader R,
Grote MJ and Martin I: Experimental and mathematical study of the
influence of growth factors on the growth kinetics of adult human
articular chondrocytes. J Cell Physiol. 204:830–838. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dorotka R, Windberger U, Macfelda K,
Bindreiter U, Toma C and Nehrer S: Repair of articular cartilage
defects treated by microfracture and a three-dimensional collagen
matrix. Biomaterials. 26:3617–3629. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rui Z, Li X, Fan J, et al: GIT1Y321
phosphorylation is required for ERK1/2- and PDGF-dependent VEGF
secretion from osteoblasts to promote angiogenesis and bone
healing. Int J Mol Med. 30:819–825. 2012.PubMed/NCBI
|
|
8
|
Pang J, Xu X, Wang X, et al:
G-protein-coupled receptor kinase interacting protein-1 mediates
intima formation by regulating vascular smooth muscle
proliferation, apoptosis, and migration. Arterioscler Thromb Vasc
Biol. 33:999–1005. 2013. View Article : Google Scholar
|
|
9
|
Menon P, Yin G, Smolock EM, Zuscik MJ, Yan
C and Berk BC: GPCR kinase 2 interacting protein 1 (GIT1) regulates
osteoclast function and bone mass. J Cell Physiol. 225:777–785.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ren Y, Yu L, Fan J, et al: Phosphorylation
of GIT1 tyrosine 321 is required for association with FAK at focal
adhesions and for PDGF-activated migration of osteoblasts. Mol Cell
Biochem. 365:109–118. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ren K, Liu F, Huang Y, et al: Periodic
mechanical stress activates integrinβ1-dependent Src-dependent
PLCγ1-independent Rac1 mitogenic signal in rat chondrocytes through
ERK1/2. Cell Physiol Biochem. 30:827–842. 2012.PubMed/NCBI
|
|
12
|
Crooke CE, Pozzi A and Carpenter GF:
PLC-gamma1 regulates fibronectin assembly and cell aggregation. Exp
Cell Res. 315:2207–2214. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Husain D, Meyer RD, Mehta M, et al: Role
of c-Cbl-dependent regulation of phospholipase Cgamma1 activation
in experimental choroidal neovascularization. Invest Ophthalmol Vis
Sci. 51:6803–6809. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hunter I, Mascall KS, Ramos JW and Nixon
GF: A phospholipase Cγ1-activated pathway regulates transcription
in human vascular smooth muscle cells. Cardiovasc Res. 90:557–564.
2011.
|
|
15
|
Kayal RA, Siqueira M, Alblowi J, McLean J,
Krothapalli N, Faibish D, Einhorn TA, Gerstenfeld LC and Graves DT:
TNF-alpha mediates diabetes-enhanced chondrocyte apoptosis during
fracture healing and stimulates chondrocyte apoptosis through
FOXO1. J Bone Miner Res. 25:1604–1615. 2010. View Article : Google Scholar
|
|
16
|
Oreffo RO: Growth factors for skeletal
reconstruction and fracture repair. Curr Opin Investig Drugs.
5:419–423. 2004.PubMed/NCBI
|
|
17
|
Stolker JM, Spertus JA, McGuire DK, et al:
Relationship between glycosylated hemoglobin assessment and glucose
therapy intensification in patients with diabetes hospitalized for
acute myocardial infarction. Diabetes Care. 35:991–993. 2012.
View Article : Google Scholar
|
|
18
|
Coutts RD, Sah RL and Amiel D: Effects of
growth factors on cartilage repair. Instr Course Lect. 46:487–494.
1997.PubMed/NCBI
|
|
19
|
Verschure PJ, Joosten LA, van der Kraan PM
and Van den Berg WB: Responsiveness of articular cartilage from
normal and inflamed mouse knee joints to various growth factors.
Ann Rheum Dis. 53:455–460. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kumei Y, Akiyama H, Hirano M, et al: Space
flight modulates signal transduction pathway of growth factor
receptors in rat osteoblasts. Biol Sci Space. 13:142–143.
1999.PubMed/NCBI
|
|
21
|
Pang J, Xu X, Getman MR, et al: G protein
coupled receptor kinase 2 interacting protein 1 (GIT1) is a novel
regulator of mitochondrial biogenesis in heart. J Mol Cell Cardiol.
51:769–776. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang S, Hisatsune C, Matsu-Ura T and
Mikoshiba K: G-protein-coupled receptor kinase-interacting proteins
inhibit apoptosis by inositol 1,4,5-triphosphate receptor-mediated
Ca2+ signal regulation. J Biol Chem. 284:29158–29169.
2009. View Article : Google Scholar : PubMed/NCBI
|