Introduction
Gastric cancer is one of the most prevalent
malignant diseases in China and is associated with a low early
diagnosis rate and a high mortality rate (1). At present, radical surgery is the
only potentially curative approach for this life-threatening
disease. However, ~50% of patients are diagnosed at an unresectable
stage due to locally far-advanced disease or distant metastasis
(2).
Although chemotherapy has enhanced the survival rate
of patients with advanced gastric cancer, the median overall
survival rate remains poor (3).
Combining the targeted agent trastuzumab with chemotherapy for the
treatment of advanced gastric cancer has proven to be superior to
chemotherapy alone and has been approved to treat human epidermal
growth factor receptor 2-positive patients with gastric cancer
(4). However, recent trials have
revealed that other targeted agents, including bevacizumab,
cetuximab and panitumumab, do not show potential for the front-line
treatment of late-stage gastric cancer (5–7).
Since the development of novel therapeutic candidates is limited,
the combination of currently available agents that have shown
theoretical or clinical efficacy may be a potential strategy for
the treatment of patients with gastric cancer.
Oxaliplatin is a third-generation platinum complex
with potent antitumor effects. Oxaliplatin has shown similar
efficacy to cisplatin in the first-line treatment of advanced
gastric cancer, as revealed by the Randomized ECF for Advanced and
Locally Advanced Esophagogastric Cancer 2 study (8). Furthermore, the side-effects
associated with oxaliplatin were moderate compared with those
associated with cisplatin, and the drug was well tolerated
(9). At present, oxaliplatin is
widely used in the palliative and adjuvant treatment of gastric
cancer (10). However, our
previous findings demonstrated that the tyrosine kinase Src was
activated following oxaliplatin exposure in gastric cancer cells,
which may serve as a potential mechanism of chemoresistance
(11).
Histone deacetylase inhibitors (HDACIs) are a group
of novel antitumor agents that target HDACs. The overexpression of
HDACs, a group of enzymes that are responsible for the modification
of lysine acetylation, is observed in various types of cancer,
including gastric cancer, and participates in the regulation of
malignant biological behaviors, including growth, resistance to
apoptosis, angiogenesis and metastasis (12,13).
Suberoylanilide hydroxamic acid (SAHA), also known as vorinostat,
is the first HDACI to be approved by the Food and Drug
Administration for the treatment of cutaneous T-cell lymphoma.
Preclinical research has also demonstrated that SAHA may show
antitumor activity in solid tumors (14,15).
Recently, a phase I trial of SAHA in combination with cisplatin and
capecitabine was conducted in patients with advanced gastric
cancer. The median overall survival time was reported to be 18
months and the toxicity was manageable (16).
The promising findings for SAHA in combination with
cisplatin suggest that SAHA may have potential in combination with
oxaliplatin for the treatment of gastric cancer. The antitumor
mechanisms of these two agents also support a potential synergistic
effect (17–19). However, to date, the combination of
oxaliplatin and SAHA in gastric cancer is yet to be investigated.
Therefore, the aim of the present study was to investigate the
antitumor effect of oxaliplatin and SAHA in gastric cancer and to
explore the potential molecular mechanisms.
Materials and methods
Cell lines and cell culture
SGC-7901 and MKN28 gastric cancer cells were
preserved in the Ruijin Hospital (Shanghai, China). The Hs746T cell
line was purchased from the American Type Culture Collection
(Manassas, VA, USA). Cells were cultured in RPMI-1640 medium with
10% fetal calf serum at 37°C and with 5% CO2.
Reagents
SAHA (S1047; Selleckchem, Houston, TX, USA) was
dissolved in dimethyl sulfoxide (DMSO) at a concentration of 200
mM. Oxaliplatin (Sanofi, Paris, France) was dissolved in 5%
dextrose solution at a concentration of 10 mg/ml. Ly2940092
(Selleckchem) and dasatinib (Selleckchem) were dissolved in DMSO at
concentrations of 20 and 100 mM, respectively. All reagents were
divided into aliquots and stored at −80°C.
Cell growth inhibition assay
A total of 5,000 cells/well were seeded onto 96-well
plates and were allowed to adhere overnight. Various concentrations
of oxaliplatin (2.5, 5, 10, 15, 20 and 25 μg/ml) or SAHA (0.5, 1,
2, 4, 6 and 8 μM) were added to the medium. DMSO solution was used
as a blank control. After 48 h of treatment, the optical density of
each well was detected using the Cell Counting Kit-8 assay and the
survival rate was calculated. In the combination treatment
experiments, the concentrations of oxaliplatin and SAHA were 5
μg/ml and 4 μM, respectively.
Colony formation assay
A total of 500 cells/well were seeded onto six-well
plates. Oxaliplatin was added to the culture medium at a final
concentration of 5 μg/ml and exposed for 3 h. The oxaliplatin was
then washed off and cells were treated with 4 μM SAHA for 24 h. For
monotherapy, the cells were incubated with either 5 μg/ml
oxaliplatin for 3 h or 4 μM SAHA for 24 h. The cells were
subsequently washed with fresh medium and allowed to grow for
between 7 and 10 days. Cell colonies were fixed using 10% neutral
formalin and stained using crystal violet. The number of colonies
was counted at a low-power field using an Olympus BX50 Microscope
(Olympus, Tokyo, Japan).
Subcutaneous xenografts
SGC-7901 cells were collected and diluted to a
concentration of 1×107 cells/ml. Twelve four-week-old
male Balb/c nude mice (Institute of Zoology Chinese Academy of
Sciences, Shanghai, China) were subcutaneously inoculated with
1×106 cells and were randomly divided into four groups
(control, oxaliplatin, SAHA and oxaliplatin plus SAHA). Treatment
commenced when the length of the tumor nodules reached 4 mm. Either
oxaliplatin (2.5 mg/kg every four days) or SAHA (50 mg/kg every two
days) monotherapy, or combination therapy was administered using
intraperitoneal injection. Intraperitoneal injection of
phosphate-buffered saline (200 μl every two days) was administered
to the control group. Mouse weight and tumor nodule size were
measured following treatment. Xenograft volume (V) was calculated
using the following formula: V = (width)2 × length/2.
The study was approved by the ethics committee of Ruijin Hospital,
Shanghai Jiaotong University School of Medicine, Shanghai,
China.
Western blot analysis
Total cell protein was extracted using
radioimmunoprecipitation assay lysis buffer (Beijing Solarbio
Science and Technology Co., Ltd, Beijing, China) after 24 h
exposure to mono- or combination therapy. Protein concentration was
determined using a DC™ protein assay (Bio-Rad, Hercules, CA, USA).
Samples containing 100 μg protein were separated using SDS-PAGE and
transferred to polyvinylidene fluoride membranes. The membranes
were blocked using 5% skimmed milk for 1 h at room temperature and
incubated overnight at 4°C with the following primary antibodies:
Anti-acetyl-histone H3, caspase-3, cleaved poly (ADP-ribose)
polymerase (PARP), phosphorylated- (p-)Akt, Akt, p-Src (all
1:1,000; Cell Signaling Technology, Inc., Danvers, MA, USA),
-B-cell lymphoma 2 (Bcl-2), Src and phosphorylated histone H2AX
(γH2AX) (all 1:1,000; Epitomics Inc., Burlingame, CA, USA). β-actin
(1:5,000; Sigma Aldrich, St. Louis, MO, USA) was used as a loading
control. The membranes were then incubated with secondary
antibodies (1:20,000; Li-Cor Biosciences, Lincoln, NE, USA) at room
temperature for 1 h. The membranes were visualized using an
infrared imaging system (Li-Cor Biosciences).
Statistical analysis
SPSS 13.0 software (SPSS, Inc., Chicago, IL, USA)
was used for the statistical analyses. Quantitative data were
analyzed using one-way analysis of variance. Factorial design
analysis was used to analyze the interaction between oxaliplatin
and SAHA. P<0.05 was considered to indicate a statistically
significant difference.
Results
Growth inhibitory effect of oxaliplatin
and SAHA in gastric cancer cells
SGC-7901, Hs746T and MKN28 gastric cancer cells were
treated with various doses of oxaliplatin or SAHA. A dose-dependent
inhibition of cell growth was observed in each treatment group.
Although the MKN28 cells were observed to be more sensitive to SAHA
than the SGC-7901 and Hs724T cells, the survival rates of the three
cell lines following treatment with 4 μM SAHA were all ~50%
(Fig. 1A). Based on these
findings, oxaliplatin and SAHA were used at concentrations of 5
μg/ml and 4 μM, respectively, in the subsequent experiments.
Oxaliplatin plus SAHA inhibits gastric
cancer cell growth in vitro
Growth inhibition and colony formation assays were
used to assess the inhibitory effect of oxaliplatin plus SAHA
combination treatment. Cell survival was observed to be
significantly impaired in the combination group compared with that
in the monotherapy groups (Fig.
1B; P<0.001). Factorial design analysis revealed a positive
interaction between oxaliplatin and SAHA in all cell lines
(between-subject effects of oxaliplatin and SAHA, P<0.001). The
colony formation assay showed that the number of colonies was
significantly lower in the combination group than that in the other
groups (Fig. 1C; P<0.001).
Oxaliplatin plus SAHA suppresses
xenograft growth in vivo
SGC-7901 gastric cancer xenografts were established
in nude mice, prior to the administration of oxaliplatin, SAHA or
combination therapy. After two weeks of treatment, tumor growth was
observed to be reduced in the mice in the mono- and combination
therapy groups, compared with that in the control group (Fig. 2). The xenograft tumor volume was
lower in the combination group than that in the oxaliplatin
(622.2±79.3 vs. 1,680.0±291.8 mm3, P=0.003) and SAHA
(622.2±79.3 vs. 1,087.0±523.8 mm3, P=0.098) groups.
Furthermore, the xenograft tumor volume in the SAHA group was lower
than that in the oxaliplatin group (1,087.0±523.8 vs. 1,680.0±291.8
mm3, P=0.044). The body weight of the mice in the four
groups was similar prior and subsequent to treatment (P>0.05;
Fig. 2).
Oxaliplatin plus SAHA enhances DNA damage
and cell apoptosis
The expression of acetyl-histone H3, γH2AX, Bcl-2,
cleaved caspase-3 and cleaved PARP was assessed following
oxaliplatin, SAHA or combination treatment using western blot
analysis (Fig. 3). Acetyl-histone
H3 expression was found to be elevated in the SAHA monotherapy and
SAHA plus oxaliplatin treatment groups, while oxaliplatin
monotherapy had no impact on the acetylation of histone H3. γH2AX
expression was observed to be significantly upregulated in the
combination group compared with that in the monotherapy groups, and
Bcl-2 expression was reduced. Cleaved caspase-3 expression was
elevated following combination treatment in Hs746T and MKN28 cells
and PARP cleavage expression was increased in Hs746T cells.
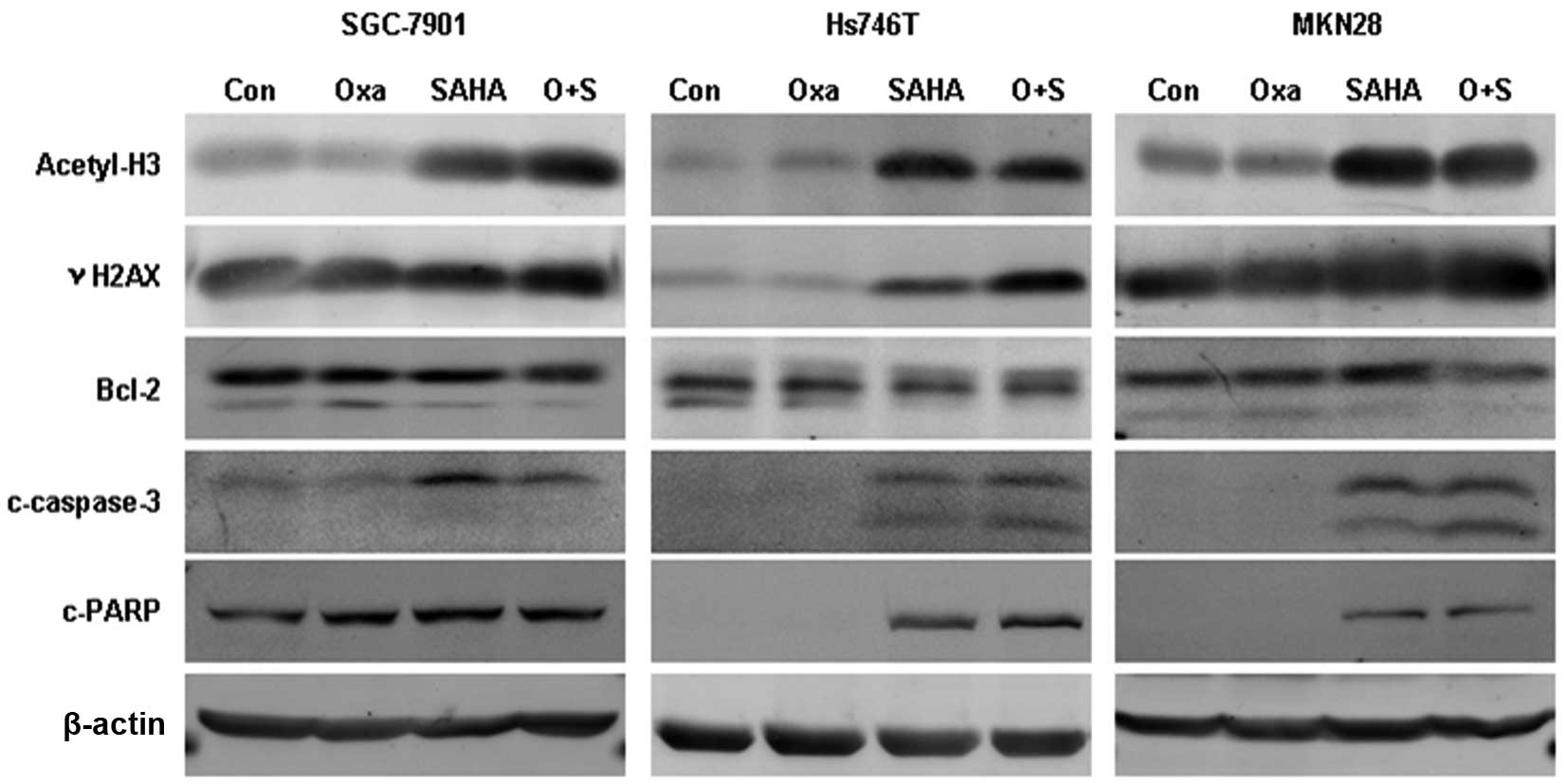 | Figure 3Expression of acetyl-H3, γH2AX, Bcl-2,
c-caspase 3 and c-PARP following treatment with Oxa and SAHA
monotherapy or combination therapy. After 24 h exposure to Oxa (5
μg/ml) and/or SAHA (4 μM) the expression of acetyl-H3, γH2AX,
Bcl-2, c-caspase 3 and c-PARP was detected using western blot
analysis. Oxa, oxaliplatin; SAHA, suberoylanilide hydroxamic acid;
acetyl-H3, acetyl-histone H3; γH2AX; phosphorylated histone H2AX;
Bcl-2; B-cell lymphoma 2; c-caspase-3, cleaved caspase-3; c-PARP,
cleaved poly (ADP-ribose) polymerase. |
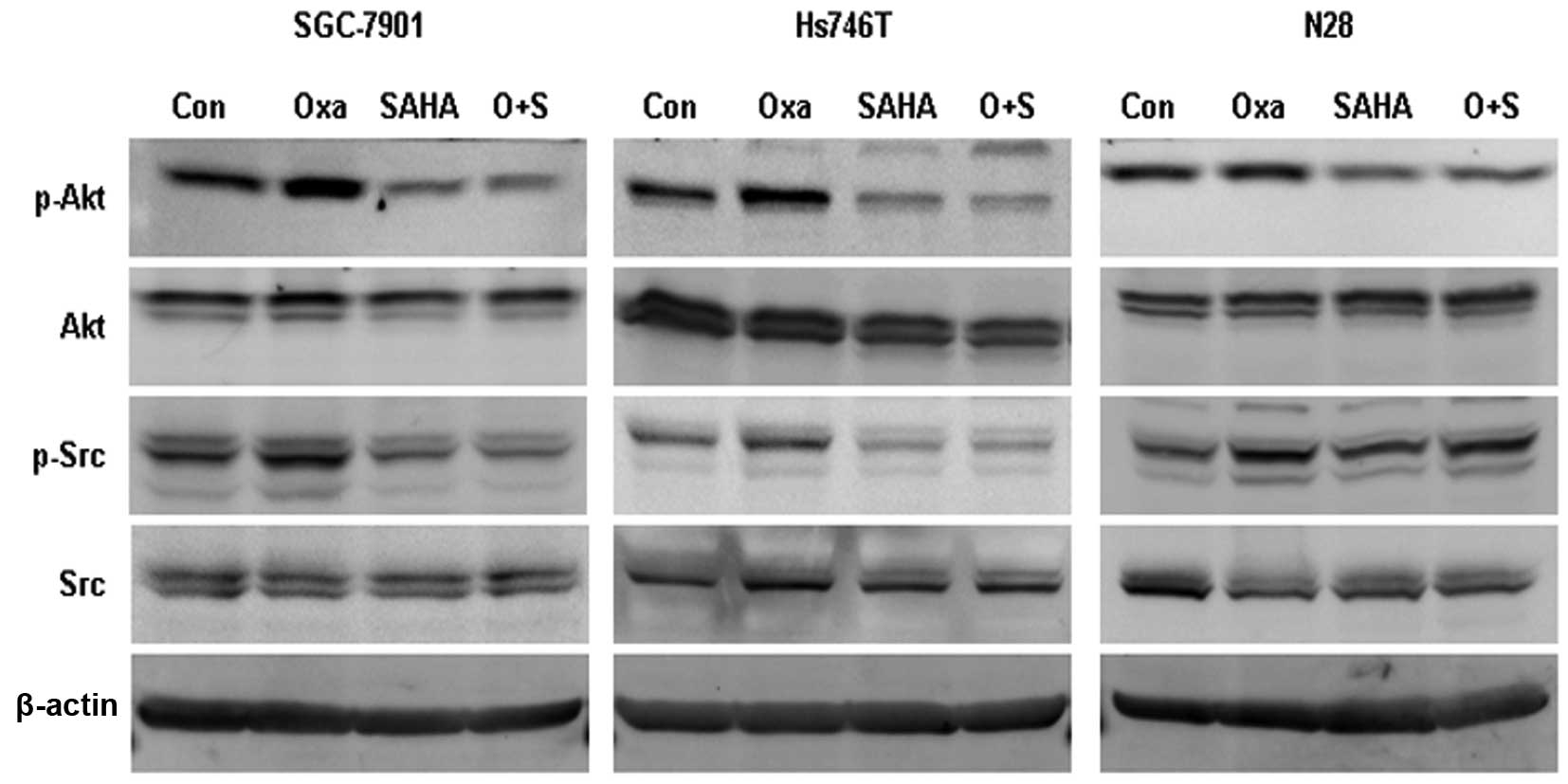
SAHA inhibits oxaliplatin-induced Src
phosphorylation
The phosphorylated forms of Src (Tyrosine 416) and
Akt (Serine 473) were observed to be elevated following oxaliplatin
exposure. However, SAHA and SAHA plus oxaliplatin treatments were
found to significantly inhibit p-Src and p-Akt expression (Fig. 4).
SAHA has been reported to attenuate Akt activation,
while its effect on Src activation remains unclear (20). In order to clarify the association
between Src and Akt activation following oxaliplatin exposure,
gastric cancer cells were treated with oxaliplatin combined with
either the phosphatidylinositide 3-kinase inhibitor Ly294002 (50
μM) or the Src inhibitor dasatinib (50 nM). Ly294002 was observed
to inhibit oxaliplatin-induced p-Akt expression, but had no impact
on oxaliplatin-induced p-Src expression. Dasatinib was found to
impair oxaliplatin-induced p-Src expression but had no effect on
oxaliplatin-induced p-Akt expression (Fig. 5A).
To investigate the interactive effect of Akt and Src
inhibition with oxaliplatin, cell survival rates were assessed in
Hs746T cells treated with oxaliplatin plus either Ly294002 or
dasatinib. Both combinations were observed to increase the
inhibition rate of Hs746T cells compared with the monotherapy.
Furthermore, the cell inhibition rate was significantly increased
in the oxaliplatin plus dasatinib group compared with that in the
oxaliplatin plus Ly294002 group (Fig.
5B; P<0.001).
Discussion
Combination therapy, including doublet/triplet
chemotherapy or chemotherapy plus targeted agents, is the primary
strategy for the front-line treatment of late-stage gastric cancer.
To optimize the efficacy and minimize the adverse events associated
with combination therapy, translational research is required to
elucidate the interactive mechanisms between different drugs. At
present, oxaliplatin is frequently used in platinum complex-based
regimes. In this era of targeted therapy, the optimal drug to be
combined with oxaliplatin is yet to be elucidated. The present
study aimed to provide preclinical evidence for agents to combine
with oxaliplatin and to investigate the interaction between
oxaliplatin and SAHA.
SAHA is a pan-HDACI that targets all the classical
HDACs, including class I, II and IV HDACs (21). Gene signature analysis performed by
Claerhout et al (22)
suggested that SAHA may be a potential drug candidate for the
treatment of gastric cancer (22).
Due to its effect on histone hyperacetylation and the modification
of chromosome structure, SAHA is considered to be a sensitizer of
DNA damage-inducing agents (23,24).
The present study showed that combining oxaliplatin with SAHA
significantly increased the inhibitory effect of oxaliplatin in
vitro and in vivo, and without significant toxicity.
This positive interaction between oxaliplatin and SAHA in gastric
cancer cells suggests that these two agents may have potential to
be used in combination in gastric cancer treatment.
As a platinum-based drug, oxaliplatin causes cell
cycle arrest and apoptosis primarily through inducing DNA damage.
Although oxaliplatin induces the formation of fewer DNA adducts
than cisplatin, the DNA damage caused by oxaliplatin is also potent
(25,26). SAHA facilitates the accessibility
of DNA damage factors; therefore, SAHA may induce
replication-dependent DNA damage (27). These findings indicate that DNA
damage may be one of the mechanisms through which oxaliplatin and
SAHA interact. Following oxaliplatin plus SAHA exposure, the
expression of γH2AX, an early marker of DNA double-strand breaks,
was significantly increased, indicating that the combination of
oxaliplatin and SAHA potentiated DNA damage.
The regulation of apoptosis is one of the mechanisms
underlying the antitumor effect of SAHA. SAHA has been reported to
regulate apoptosis by decreasing the expression of Bcl-2 and
Bcl-extra large, and increasing that of Bcl-2-associated X protein
and Bcl-2 homologous antagonist killer. New et al (28) and Thompson et al (29) found that exogenous expression of
Bcl-2 impaired SAHA-induced apoptosis in diffuse large B-lymphoma
cells (28,29). In the present study, Bcl-2
expression was not changed in the gastric cancer cells in the SAHA
or oxaliplatin monotherapy groups, but was reduced in the
combination treatment group. However, following oxaliplatin and
SAHA doublet treatment, the expression of cleaved caspase-3 and
cleaved PARP was found to be increased, indicating that apoptosis
was potentiated
In addition to its effects on apoptosis, SAHA was
found to reverse the oxaliplatin-induced Src activation. The
effects of SAHA on Src activation have been rarely reported.
Trichostatin A (TSA) and butyrate, two common HDACIs, have been
found to inhibit Src expression in colon cancer cell lines
(30). However, in the present
study, SAHA was not observed to significantly change total Src
expression, indicating that the regulation of relative signaling
pathways may contribute to this phenomenon.
HDACIs, including SAHA, have been reported to
inhibit Akt activation. TSA has been found to increase the
association between protein phosphatase 1 (PP1) and Akt through the
disassembly of the HDAC/PP1 complex (20). In addition, cross-talk between Src
and Akt has been reported in cancer cells (31). In present study, SAHA was observed
to induce the suppression of Akt activation. However, the
inhibitory effect of SAHA on Src activation appeared to be
independent of its effect on Akt in the three gastric cancer cell
lines.
The kinases Src and Akt are important for tumor cell
survival and growth (32,33). We previously reported that Src
phosphorylation was upregulated by oxaliplatin in gastric cancer
cells, and that the Src inhibitor dasatinib showed a significant
synergic effect with oxaliplatin (11). The inhibition of Akt activation has
also been reported to potentiate the antitumor effect of
oxaliplatin (34). In the present
study, combining oxaliplatin with dasatinib or Ly294002 inhibited
Hs746T cell growth compared with monotherapy, and oxaliplatin plus
dasatinib showed a more potent efficacy than oxaliplatin plus
Ly294002. These findings indicate that Src activation may have an
important role in the interaction between oxaliplatin and SAHA.
In conclusion, the combination of oxaliplatin with
SAHA potentiated its inhibitory effect in gastric cancer cells. The
reversal of oxaliplatin-induced Src phosphorylation may be one of
mechanisms by which SAHA enhances the efficacy of oxaliplatin. The
present study has identified potential drug combinations for
chemotherapy in gastric cancer, which may warrant investigation in
clinical trials. The mechanism by which SAHA suppresses Src
activation should be investigated further.
Acknowledgements
This study was supported by grants from the National
Science Foundation of China (nos. 81372645 and 30801371), Shanghai
Natural Science Foundation from the Municipal Government (no.
13ZR1425900), Shanghai Jiao Tong University School of Medicine
Science and Technology Foundation (13XJ10035), the Fong Shu Fook
Tong Foundation and by Doctoral Innovation Fund Projects from
Shanghai Jiaotong University School of Medicine (no.
BXJ201317).
References
|
1
|
Zhao P, Dai M, Chen W and Li N: Cancer
trends in China. Jpn J Clin Oncol. 40:281–285. 2010. View Article : Google Scholar
|
|
2
|
Hartgrink HH, Jansen EP, van Grieken NC
and van de Velde CJ: Gastric cancer. Lancet. 374:477–490. 2009.
View Article : Google Scholar
|
|
3
|
Cervantes A, Roda D, Tarazona N, et al:
Current questions for the treatment of advanced gastric cancer.
Cancer Treat Rev. 39:60–67. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bang YJ, Van Cutsem E, Feyereislova A, et
al: ToGA Trial Investigators: Trastuzumab in combination with
chemotherapy versus chemotherapy alone for treatment of
HER2-positive advanced gastric or gastro-oesophageal junction
cancer (ToGA): a phase 3, open-label, randomised controlled trial.
Lancet. 376:687–697. 2010. View Article : Google Scholar
|
|
5
|
Ohtsu A, Shah MA, Van Cutsem E, et al:
Bevacizumab in combination with chemotherapy as first-line therapy
in advanced gastric cancer: a randomized, double-blind,
placebo-controlled phase III study. J Clin Oncol. 29:3968–3976.
2011. View Article : Google Scholar
|
|
6
|
Lordick F, Kang YK, Chung HC, et al:
Arbeitsgemeinschaft Internistische Onkologie and EXPAND
Investigators: Capecitabine and cisplatin with or without cetuximab
for patients with previously untreated advanced gastric cancer
(EXPAND): a randomised, open-label phase 3 trial. Lancet Oncol.
14:490–499. 2013. View Article : Google Scholar
|
|
7
|
Waddell T, Chau I, Cunningham D, et al:
Epirubicin, oxaliplatin, and capecitabine with or without
panitumumab for patients with previously untreated advanced
oesophagogastric cancer (REAL3): a randomised, open-label phase 3
trial. Lancet Oncol. 14:481–489. 2013. View Article : Google Scholar
|
|
8
|
Cunningham D, Starling N, Rao S, et al:
Upper Gastrointestinal Clinical Studies Group of the National
Cancer Research Institute of the United Kingdom: Capecitabine and
oxaliplatin for advanced esophagogastric cancer. N Engl J Med.
358:36–46. 2008. View Article : Google Scholar
|
|
9
|
Rabik CA and Dolan ME: Molecular
mechanisms of resistance and toxicity associated with platinating
agents. Cancer Treat Rev. 33:9–23. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bang YJ, Kim YW, Yang HK, et al: CLASSIC
trial investigators; Adjuvant capecitabine and oxaliplatin for
gastric cancer after D2 gastrectomy (CLASSIC): a phase 3
open-label, randomised controlled trial. Lancet. 379:315–321. 2012.
View Article : Google Scholar
|
|
11
|
Shi M, Lou B, Ji J, et al: Synergistic
antitumor effects of dasatinib and oxaliplatin in gastric cancer
cells. Cancer Chemother Pharmacol. 72:35–44. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Weichert W, Röske A, Gekeler V, et al:
Association of patterns of class I histone deacetylase expression
with patient prognosis in gastric cancer: a retrospective analysis.
Lancet Oncol. 9:139–148. 2008. View Article : Google Scholar
|
|
13
|
Barneda-Zahonero B and Parra M: Histone
deacetylases and cancer. Mol Oncol. 6:579–589. 2012. View Article : Google Scholar
|
|
14
|
Mann BS, Johnson JR, Cohen MH, et al: FDA
approval summary: vorinostat for treatment of advanced primary
cutaneous T-cell lymphoma. Oncologist. 12:1247–1252. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Carew JS, Giles FJ and Nawrocki ST:
Histone deacetylase inhibitors: mechanisms of cell death and
promise in combination cancer therapy. Cancer Lett. 269:7–17. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yoo C, Ryu MH, Na YS, et al: Phase I and
pharmacodynamic study of vorinostat combined with capecitabine and
cisplatin as first-line chemotherapy in advanced gastric cancer.
Invest New Drugs. 32:271–278. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Raymond E, Faivre S, Chaney S, et al:
Cellular and molecular pharmacology of oxaliplatin. Mol Cancer
Ther. 1:227–235. 2002.
|
|
18
|
Petruccelli LA, Dupéré-Richer D,
Pettersson F, et al: Vorinostat induces reactive oxygen species and
DNA damage in acute myeloid leukemia cells. PLoS One. 6:e209872011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang C, Richon V, Ni X, et al: Selective
induction of apoptosis by histone deacetylase inhibitor SAHA in
cutaneous T-cell lymphoma cells: relevance to mechanism of
therapeutic action. J Invest Dermatol. 125:1045–1052. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen CS, Weng SC, Tseng PH, et al: Histone
acetylation-independent effect of histone deacetylase inhibitors on
Akt through the reshuffling of protein phosphatase 1 complexes. J
Biol Chem. 280:38879–38887. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bolden JE, Peart MJ and Johnstone RW:
Anticancer activities of histone deacetylase inhibitors. Nat Rev
Drug Discov. 5:769–784. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Claerhout S, Lim JY, Choi W, et al: Gene
expression signature analysis identifies vorinostat as a candidate
therapy for gastric cancer. PLoS One. 6:e246622011. View Article : Google Scholar
|
|
23
|
Diyabalanage HV, Granda ML and Hooker JM:
Combination therapy: histone deacetylase inhibitors and
platinum-based chemotherapeutics for cancer. Cancer Lett. 329:1–8.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rikiishi H, Shinohara F, Sato T, et al:
Chemosensitization of oral squamous cell carcinoma cells to
cisplatin by histone deacetylase inhibitor, suberoylanilide
hydroxamic acid. Int J Oncol. 30:1181–1188. 2007.PubMed/NCBI
|
|
25
|
Woynarowski JM, Faivre S, Herzig MC, et
al: Oxaliplatin-induced damage of cellular DNA. Mol Pharmacol.
58:920–927. 2000.PubMed/NCBI
|
|
26
|
Faivre S, Chan D, Salinas R, et al: DNA
strand breaks and apoptosis induced by oxaliplatin in cancer cells.
Biochem Pharmacol. 66:225–237. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Conti C, Leo E, Eichler GS, et al:
Inhibition of histone deacetylase in cancer cells slows down
replication forks, activates dormant origins, and induces DNA
damage. Cancer Res. 70:4470–4480. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
New M, Olzscha H and La Thangue NB: HDAC
inhibitor-based therapies: can we interpret the code? Mol Oncol.
6:637–656. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Thompson RC, Vardinogiannis I and Gilmore
TD: The sensitivity of diffuse large B-cell lymphoma cell lines to
histone deacetylase inhibitor-induced apoptosis is modulated by
BCL-2 family protein activity. PLoS One. 8:e628222013. View Article : Google Scholar
|
|
30
|
Hirsch CL, Smith-Windsor EL and Bonham K:
Src family kinase members have a common response to histone
deacetylase inhibitors in human colon cancer cells. Int J Cancer.
118:547–554. 2006. View Article : Google Scholar
|
|
31
|
Jiang T and Qiu Y: Interaction between Src
and a C-terminal proline-rich motif of Akt is required for Akt
activation. J Biol Chem. 278:15789–15793. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Summy JM and Gallick GE: Treatment for
advanced tumors: SRC reclaims center stage. Clin Cancer Res.
12:1398–1401. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hsieh AC, Truitt ML and Ruggero D:
Oncogenic AKTivation of translation as a therapeutic target. Br J
Cancer. 105:329–336. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu J, Fu XQ, Zhou W, et al: LY294002
potentiates the anti-cancer effect of oxaliplatin for gastric
cancer via death receptor pathway. World J Gastroenterol.
17:181–190. 2011. View Article : Google Scholar : PubMed/NCBI
|