Introduction
Ischemic postconditioning (IPoC), a short series of
repetitive cycles of brief reperfusion and ischemia performed
immediately following sustained ischemia, has been found to reduce
myocardial ischemia reperfusion injury (1). The cardioprotection of IPoC has been
proved in several animal models and even in the human heart
(2–4). Although existing data in the
literature remain contradictory, the majority of studies have
demonstrated that hypercholesterolemia interferes with the
cardioprotective effect of IPoC (5–8).
However, the mechanism underlying the attenuation of the
cardioprotective effects of IPoC in hypercholesterolemic conditions
is not well understood.
Previously, several studies have demonstrated that
hypercholesterolemia abrogates cardioprotection of IPoC or
sevoflurane-induced preconditioning involved in inhibiting the
activation of phosphatidylinositol 3-kinase (PI3K)/Akt/endothelial
nitric oxide synthase (eNOS) pathway (9–11).
Furthermore, pravastatin, a 3-hydroxy-3-methylglutaryl-CoA
(HMG-CoA) reductase inhibitor, has been proved to be able to
restore the cardioprotection of IPoC in hypercholesterolemic states
by upregulating the PI3K/Akt/eNOS pathway, which is possibly
independent of its lipid-lowing effect (9).
A number of studies have demonstrated that fasudil,
a Rho-kinase inhibitor, had beneficial effects in diverse
cardiovascular diseases, including ischemic heart disease,
atherosclerosis, myocardial hypertrophy and heart failure (12–15).
Furthermore, fasudil has been reported to lead to the activation of
the PI3K/Akt/eNOS pathway, resulting in preconditioning and
postconditioning against myocardial ischemia reperfusion injury
(16–18). Therefore, it was hypothesized that
fasudil may restore the cardioprotection of IPoC in the presence of
hypercholesterolemia.
Based on this hypothesis, the present study examined
whether the administration of fasudil shortly prior to ischemia may
restore the cardioprotection of IPoC in the presence of
hypercholesterolemia. Furthermore, in order to determine the
potential underlying mechanisms, the myocardial NO content and
Rho-kinase activity, as well as the activation of PI3K/Akt/eNOS
pathway in this process, were assessed.
Materials and methods
Animals
A total of 105 male Wistar rats purchased from the
Center of Experimental Animals, China Medical University (Shenyang,
Liaoning, China), weighing 200±10 g were used in the present study.
All of the animals used in this study were treated in accordance
with the Guide for the Care and Use of Laboratory Animals,
published by the National Institutes of Health (NIH). The study
procedure was approved by the institutional Ethics Committee of
China Medical University (Shenyang, China).
Drugs
Fasudil was purchased from Chase Sun Pharmaceutical
Company (Tianjin, China). Wortmannin, L-NAME and 2,3,5-triphenyl
tetrazoliumchloride (TTC) were purchased from Sigma (St. Louis, MO,
USA).
Induction of experimental
hypercholesterolemia
Prior to the initiation of the 8-week feeding
period, the blood samples were obtained from the rats’ vena
caudalis for determination of the plasma levels of total
cholesterol (TC), low density lipoprotein (LDL) and high density
lipoprotein (HDL) using commercial kits (Total Cholesterol Assay
kit - Fluoro Cholesterol; Cell Technology, Mountain View, CA, USA;
Cholesterol LDL direct; Biosystems S.A., Barcelona, Spain; and
Cholesterol HDL direct, Biosystems S.A). All of the animals were
fed with a diet enriched with 1.5% cholesterol, 5% egg yolk powder,
10% lard, 0.5% sodium cholate, 3% sugar and 80% normal feed for 8
weeks, and this formula was modified as previously described
(19). Following the 8-week
feeding period, the blood samples were obtained from the rats’ vena
caudalis again for determination of serum lipid in order to judge
the success of hypercholesterolemic models.
Heart preparation
The rats were anesthetized with an intraperitoneal
injection of pentobarbital sodium (100 mg/kg). Heparin (1,500
IU/kg) was administered intravenously to prevent intracoronary clot
formation. The heart was rapidly excised and immediately immersed
in ice-cold heparinized modified Krebs-Henseleit buffer containing
(in per mmol) 127 NaCl, 17.7 NaHCO3, 5.1 KCl, 1.5
CaCl2, 1.26 MgCl2 and 11D-glucose (pH 7.4).
The heart was mounted on a Langendorff-perfusion apparatus and
retrogradely perfused through the aorta with recirculating buffer
saturated with 95% O2-5% CO2 at 37°C. The
heart was maintained in a thermostatic chamber at 37°C. Perfusion
was maintained at a constant pressure of 75 mmHg. The fluid-filled
latex balloon was inserted in the left ventricle (LV) via the left
atrium for pressure measurement. The balloon was connected to a
pressure transducer and inflated to an initial LV end-diastolic
pressure between 8 and 10 mmHg.
Experimental procedure
The rats were further divided into seven groups with
15 animals/group. In all of the groups, the isolated rat hearts
were perfused with a K-H solution (127 mM NaCl, 17.7 mM
NaHCO3, 5.1 mM KCl, 1.5 mM CaCl2, 1.26 mM
MgCl2 and 11mM D-glucose, pH 7.4) and allowed 10 min of
stabilization. Next, all of the isolated rat hearts were subjected
to 30 min global ischemia and 120 min reperfusion.
In the control group, the isolated rat hearts were
subjected to 30 min global ischemia and 120 min reperfusion. For
the ischemic postconditioning group (IPoC group): the isolated rat
hearts were subject to ischemia for 30 min, followed by six cycles
of reperfusion and ischemia, both with equal lengths of 10 sec and
reperfusion for 120 min. In the fasudil preconditioning group (FD
group), the isolated rat hearts were perfused with K-H solution
containing 1 μM dose fasudil 15 min prior to ischemia. In the
ischemic postconditioning+fasudil group (IPoC+FD group), 1 μM
fasudil was added to the perfusate for 15 min prior to ischemia,
then 30 min ischemia was followed by six cycles of reperfusion and
ischemia, both with equal lengths of 10 sec and reperfusion for 120
min.
To investigate whether a higher concentration of
fasudil alone was able to restore cardioprotection in
hypercholesterolemic rat heart, an additional group with 10 μM
fasudil treatment prior ischemia was also established. In the high
dose fasudil preconditioning group (FD2 group), the isolated rat
hearts were perfused with K-H solution containing 10 μM fasudil 15
min prior to ischemia
To further determine whether the PI3K/Akt/eNOS
pathway participated in regulating fasudil restored
cardioprotection of IPoC in the state of hypercholesterolemia, two
more groups were added to the experimental studies, with PI3K
specific inhibitor wortmannin and eNOS specific inhibitor L-NAME
treatment. In the ischemic postconditioning+fasudil+wortmannin
group (IPoC+FD+wortmannin group), wortmannin at the dose of 30 μM
was administrated immediately following IPoC. The ischemic
postconditioning and fasudil preconditioning were performed as
described above. In the ischemic postconditioning+fasudil+L-NAME
group (IPoC+FD+L-NAME group), L-NAME at the dose of 30 μM was
administrated immediately following IPoC. Ischemic postconditioning
and fasudil preconditioning were conducted as described above.
Hemodynamic monitoring
The homodynamic assessment included heart rate (HR),
left ventricular developed pressure (LVDP), positive first order
derivative of ventricular pressure (+dp/dt) and negative first
order derivative of ventricular pressure (−dp/dt). These parameters
were continuously monitored throughout the experimental procedures.
The HR, LVDP, +dp/dt and −dp/dt were sampled and digitally
processed via a homodynamic system (BIOPAC MP150; BIOPAC systems,
Goleta, CA, USA).
Measurement of infarct size
The infarct size was determined as previously
described (20). Briefly,
following 2 h reperfusion, the hearts were harvested and the LVs
were sectioned from the apex to the base into 2–3 mm sections,
following incubation for 20 min at 37°C in 1% triphenyltetrazolium
chloride (TTC). The unstained tissue was carefully separated from
the stained tissue by an independent observer. While the unstained
tissue represented the dead cells, the stained tissue represented
the viable cells. The unstained mass was expressed as a percentage
of total LV mass. The total LV mass also corresponded to the risk
area because a global ischemia was induced.
Measurement of myocardial apoptosis
At the end of 2 h reperfusion, the heart was removed
as described above. Cardiomyocyte apoptosis was detected using an
In Situ Cell Death Detection kit (Roche, South San Francisco, CA,
USA) according to the manufacturer’s instructions. Briefly, the
tissue sections were washed in PBS and then fixed in 4%
paraformaldehyde solution prior to incubation in 20 μg/ml
proteinase K for 10 min. Following being washed in PBS for three
times, the tissue sections were incubated with terminal
deoxynucleotidyl transferase enzyme (TUNEL) in a humidified chamber
at 37°C for 60 min for incorporation of the biotinylated
nucleotides at the 3′-OH DNA ends. The reaction was terminated by
transferring the slides to a 2× sodium citrate saline solution.
Endogenous peroxidase activity was quenched by incubation in 0.3%
hydrogen peroxide. Finally, streptavidin horseradish peroxidase was
bound to the biotinylated nucleotides and the peroxidase activity
was demonstrated in each section by the application of a stable
chromogen diaminobenzidine. In this technique, the apoptotic nuclei
are stained dark brown. The sections were counter stained with
hematoxylin for total nuclei. Three sections from each myocardial
sample were randomly selected and ten microscopic fields (Olympus
BX51 microscope; Olympus, Tokyo, Japan) per section were evaluated
by two independent blind observers. In each field, the nuclei were
counted and the percentage of TUNEL-positive nuclei was
calculated.
Measurements of NO content
After 30 min of global ischemia followed by 120 min
of reperfusion, the hearts were removed rapidly from the
Langendorff apparatus and homogenized. The content of NO was
measured using Nitric Oxide assay kit (Nanjing Jiancheng
Bioengineering Institute, Nanjing, China) according to the
manufacturer’s instructions.
Western blotting
At the end of 30 min reperfusion, the LVs were
homogenized in a lysis buffer [(in mmol/l) 10 Tris-HCl, 20
ortho-phosphate, 1 EGTA, 1 EDTA, 2 Na3VO4, 1
phenylmethylsulfonyl fluoride; pH 7.4]. Following sonication, the
lysates were centrifuged, the proteins were separated by
electrophoresis on SDS-PAGE and transferred onto
polyvinylidenedifluoride-plus membranes. The membranes were blocked
with 5% skimmed milk followed by incubation overnight at 4°C with
the antibodies: MYPT-1 (1:300; Abcam); phospho-MYPT-1 (at
Ser853,1:300; Abcam, Hong Kong, China); Akt (1:200; Santa Cruz
Biotechnology Inc., Santa Cruz, CA, USA); phospho-Akt (at Ser473,
1:200; Santa Cruz Biotechnology, Inc.); eNOS (1:200; Santa Cruz
Biotechnology Inc.); phospho-eNOS (at Ser1177, 1:200; Santa Cruz
Biotechnology Inc.). Following incubation, the membranes were
washed three times with 0.1% Tween-20 for 15 min and incubated with
horseradish peroxidase for 2 h. The levels of phosphorylated
proteins were normalized to their total protein levels. The
relative densitometry was performed using a computerized software
package (NIH Image 1.63 software).
Statistical analysis
The data are expressed as the mean ± standard
deviation values. The statistical analysis was performed using
Sigma Stat software version 3.5 (Systat software). The differences
between the groups were evaluated using one-way analysis of
variance, followed by Student-Newman-Keuls post hoc test. P<0.05
were considered to indicate a statistically significant
difference.
Results
The levels of plasma lipids
Table I summarizes
the average values for TC, HDL and LDL in the plasma of animals
prior to and following the 8 week cholesterol-enriched diet. A
significant increase (P<0.05) in the average TC and LDL values
were observed in the animals fed with the cholesterol-enriched diet
as compared with the control group. There was no significant
difference in the value of HDL between the cholesterol-enriched
diet group and the control group (P>0.05).
 | Table IBiochemical analysis (mean ± standard
deviation; n=40/group). |
Table I
Biochemical analysis (mean ± standard
deviation; n=40/group).
| Total cholesterol
(mg/dl) | HDL-C (mg/dl) | LDL-C (mg/dl) |
|---|
| Before
cholesterol-enriched diet fed | 48.7±6.3 | 40.3±4.1 | 14.2±4.5 |
| After
cholesterol-enriched diet fed | 227.1±7.6a | 43.4±5.1 | 56.5±5.3a |
Hemodynamic changes
Table II
demonstrates the values of HR, LVDP, +dp/dt and −dp/dt at baseline
and different times of reperfusion. There were no significant
differences among the groups in HR, LVDP, +dp/dt and −dp/dt at
baseline. No significant differences in HR, LVDP, +dp/dt and −dp/dt
were observed in the IPoC and FD groups compared with the control
group (P>0.05). However, IPoC+FD group significantly increased
the values of HR, LVDP, +dp/dt and −dp/dt at different times of
reperfusion compared with the control group (P<0.05).
Furthermore, high doses of fasudil treatment alone (FD2 group) also
improved the values of HR, LVDP, +dp/dt and −dp/dt at different
times of reperfusion compared with the control group
(P<0.05).
| Table IIHR, +dp/dt, −dp/dt and LVDP prior to
and during reperfusion. |
Table II
HR, +dp/dt, −dp/dt and LVDP prior to
and during reperfusion.
| Time | Baseline | R-10 | R-20 | R-60 |
|---|
| HR |
| Control | 246±23 | 150±18 | 174±19 | 160±24 |
| IPoC | 257±20 | 145±26 | 165±23 | 158±15 |
| FD | 245±17 | 158±22 | 178±27 | 164±22 |
| IPoC+FD | 250±18 | 176±17a | 198±12a | 188±16a |
| FD2 | 255±16 | 173±21a | 194±15a | 185±12a |
|
IPoC+FD+wortmannin | 247±21 | 141±26 | 159±23 | 149±25 |
|
IPoC+FD+L-NAME | 251±22 | 149±22 | 172±22 | 166±17 |
| LVDP (mm Hg) |
| Control | 96±6.4 | 35±5.4 | 39±5.1 | 34±3.2 |
| IPoC | 94±6.5 | 32±7.3 | 38±4.5 | 33±4.3 |
| FD | 93±6.6 | 36±7.9 | 37±6.0 | 32±3.6 |
| IPoC+FD | 98±7.5 | 47±5.9a | 51±7.7a | 49±3.9a |
| FD2 | 94±6.9 | 44±3.7a | 49±4.2a | 46±3.4a |
|
IPoC+FD+wortmannin | 96±4.5 | 34±5.4 | 35±5.1 | 33±3.8 |
|
IPoC+FD+L-NAME | 95±7.3 | 32±7.7 | 37±6.5 | 31±6.3 |
| +dp/dt(mmHg/s) |
| Control | 2,322±233 | 1,589±103 | 1,622±208 | 1,362±198 |
| IPoC | 2,318±187 | 1,466±259 | 1,589±223 | 1,284±134 |
| FD | 2,472±274 | 1,546±202 | 1,666±244 | 1,354±143 |
| IPoC+FD | 2,312±233 | 1,742±107a | 1,832±161a | 1,662±168a |
| FD2 | 2,298±177 | 1,746±159a | 1,889±217a | 1,584±114a |
|
IPoC+FD+wortmannin | 2,402±244 | 1,536±207 | 1,566±287 | 1,454±203 |
|
IPoC+FD+L-NAME | 2,231±262 | 1,489±301 | 1,684±212 | 1,335±158 |
| −dp/dt(mmHg/s) |
| Control | 1,576±222 | 978±178 | 1,302±117 | 1,138±156 |
| IPoC | 1,578±207 | 939±167 | 1,271±189 | 1,079±134 |
| FD | 1,508±190 | 989±312 | 1,308±202 | 1,136±234 |
| IPoC+FD | 1,590±156 | 1,146±142a | 1,498±145a | 1,378±148a |
| FD2 | 1,576±222 | 1,058±168a | 1,456±177a | 1,262±176a |
|
IPoC+FD+wortmannin | 1,595±137 | 1,009±187 | 1,278±182 | 1,169±144 |
|
IPoC+FD+L-NAME | 1,601±165 | 1,018±198 | 1,274±237 | 1,156±159 |
Infarct size measurement
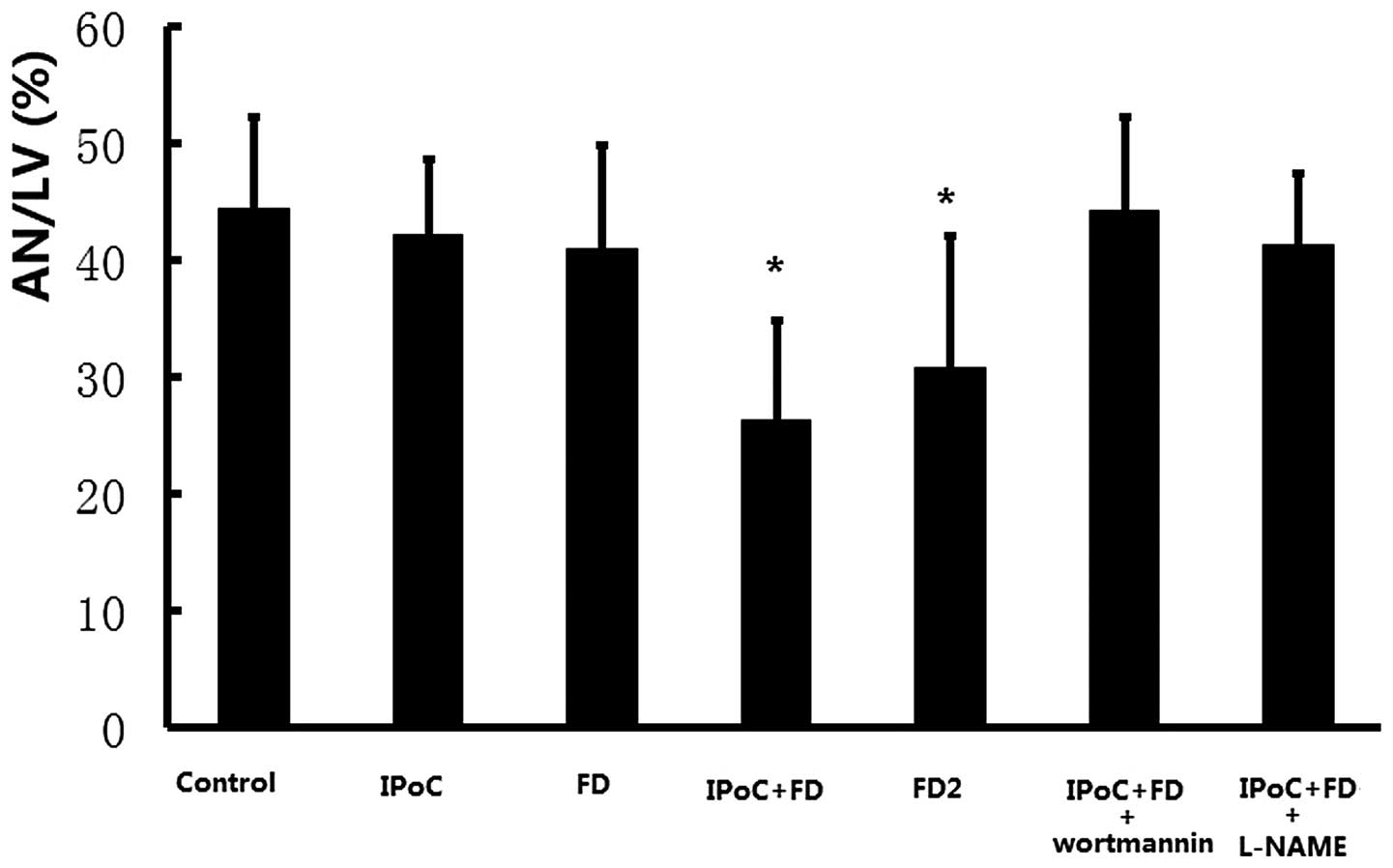
As demonstrated in Fig.
1, no significant decrease in infarct size was observed in IPoC
group and FD group as compared with the control group (42.3±6.5 and
40.9±8.9%, respectively, vs. 44.4±7.9%, P>0.05). However, the
IPoC+FD group had a significantly reduced infarct size as compared
with the control group (26.3±8.5 vs. 44.4±7.9%, P<0.05),
suggesting that low dose fasudil preconditioning restored the
cardioprotection of IPoC in hypercholesterolemic conditions. In
addition, a high dose of fasudil treatment alone (FD2 group) also
reduced infarct size as compared with the control group (30.8±11.3
vs. 44.4±7.9%, P<0.05).
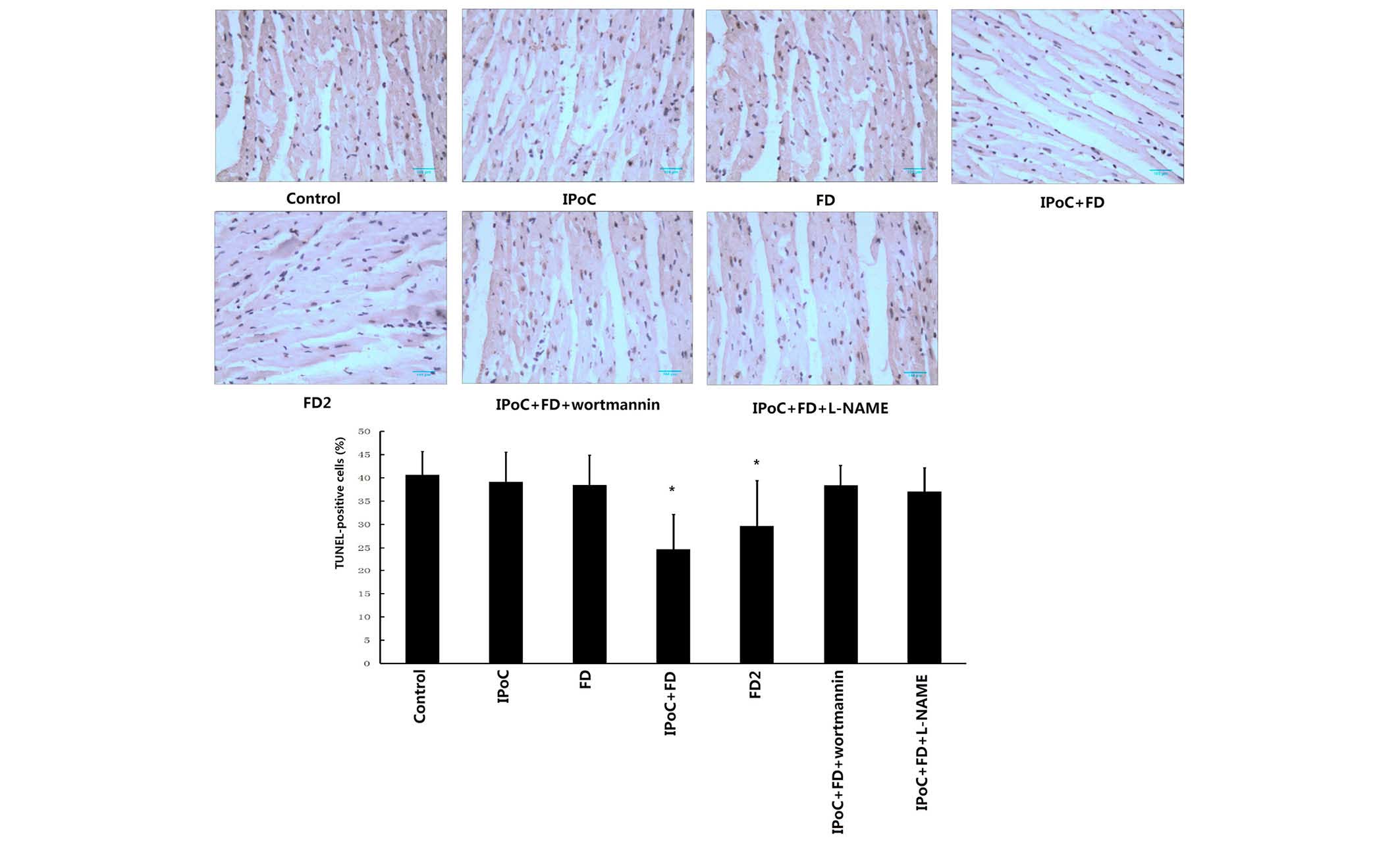
TUNEL staining for apoptosis
The percentage of apoptotic cardiomyocytes of all of
the experimental groups is demonstrated in Fig. 2. The IPoC+FD group and FD2 group
significantly reduced cardiomyocyte apoptosis as compared with the
control group (24.7±4.5 and 29.6±9.8%, respectively, vs. 40.6±5.0%,
P<0.05). However, IPoC and low dose of fasudil treatment alone
failed to exert the anti-apoptosis effect, as evidenced by no
significant decrease in the percentage of cardiomyocyte apoptosis
in IPoC and FD groups, as compared with the control group (39.1±6.4
and 38.5±6.4%, respectively, vs. 40.6±5.0%, P>0.05).
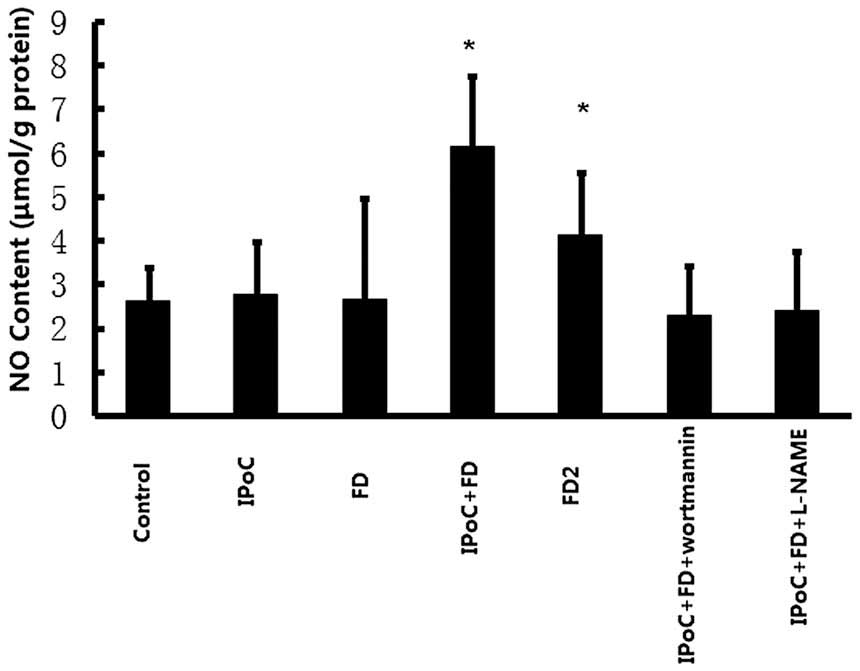
Effects on NO content in rat
myocardium
As revealed in Fig.
3, the content of NO in rat myocardium was significantly
increased in the IPoC+FD group, as compared with the control group
(6.15±1.60 vs. 2.60±0.78, P<0.05), but the content of NO was not
increased in the IPoC and FD groups (2.77±1.20 and 2.65±2.31,
respectively, vs. 2.60±0.78, P>0.05). High dose fasudil
preconditioning also markedly enhanced the content of NO in the rat
myocardium as compared with the control group (4.12±1.43 vs.
2.60±0.78, P<0.05).
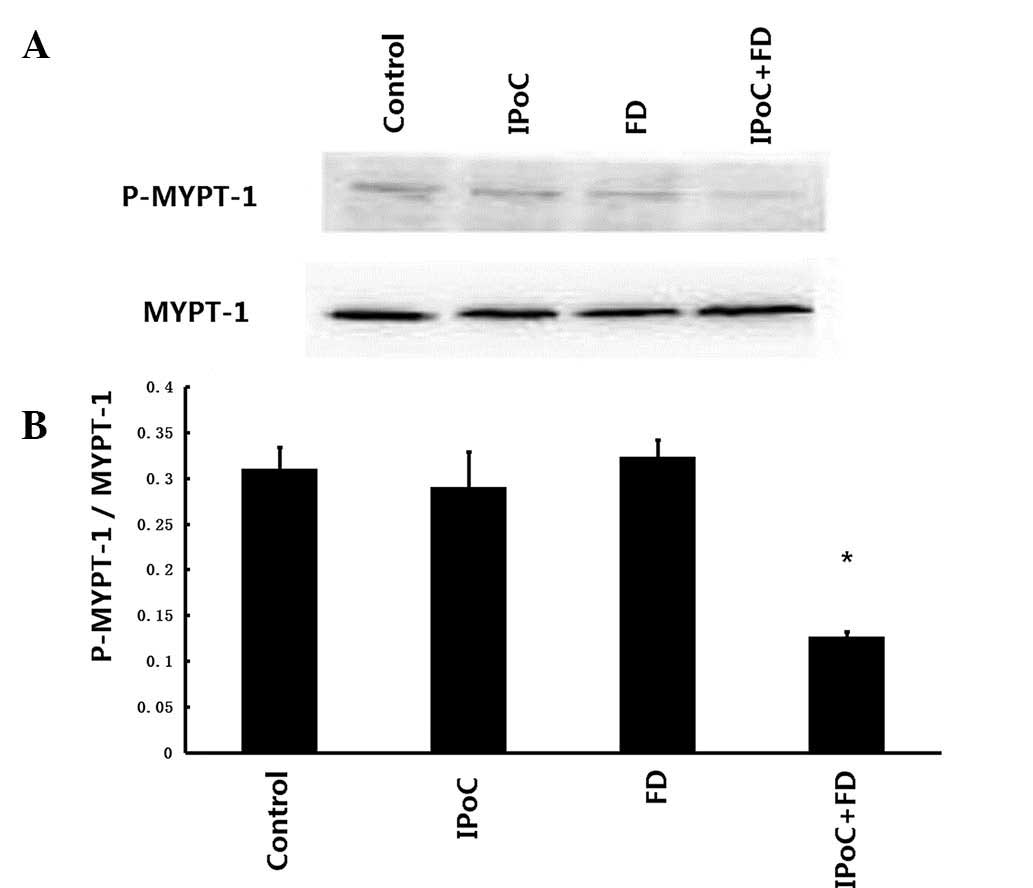
Activity of Rho-kinase
The Rho-kinase activity was assessed by examining
the phosphorylation of MYPT-1, a well-established Rho-kinase
specific substrate. As demonstrated in Fig. 4A and B, IPoC+FD treatment resulted
in a significant reduction in MYPT-1 phosphorylation as compared
with the control group (0.126±0.006 vs. 0.310±0.024, P<0.05),
whereas no significant decrease in MYPT-1 phosphorylation was
revealed in the IPoC and FD groups (0.289±0.039 and 0.323±0.019,
respectively, vs. 0.310±0.024, P>0.05).
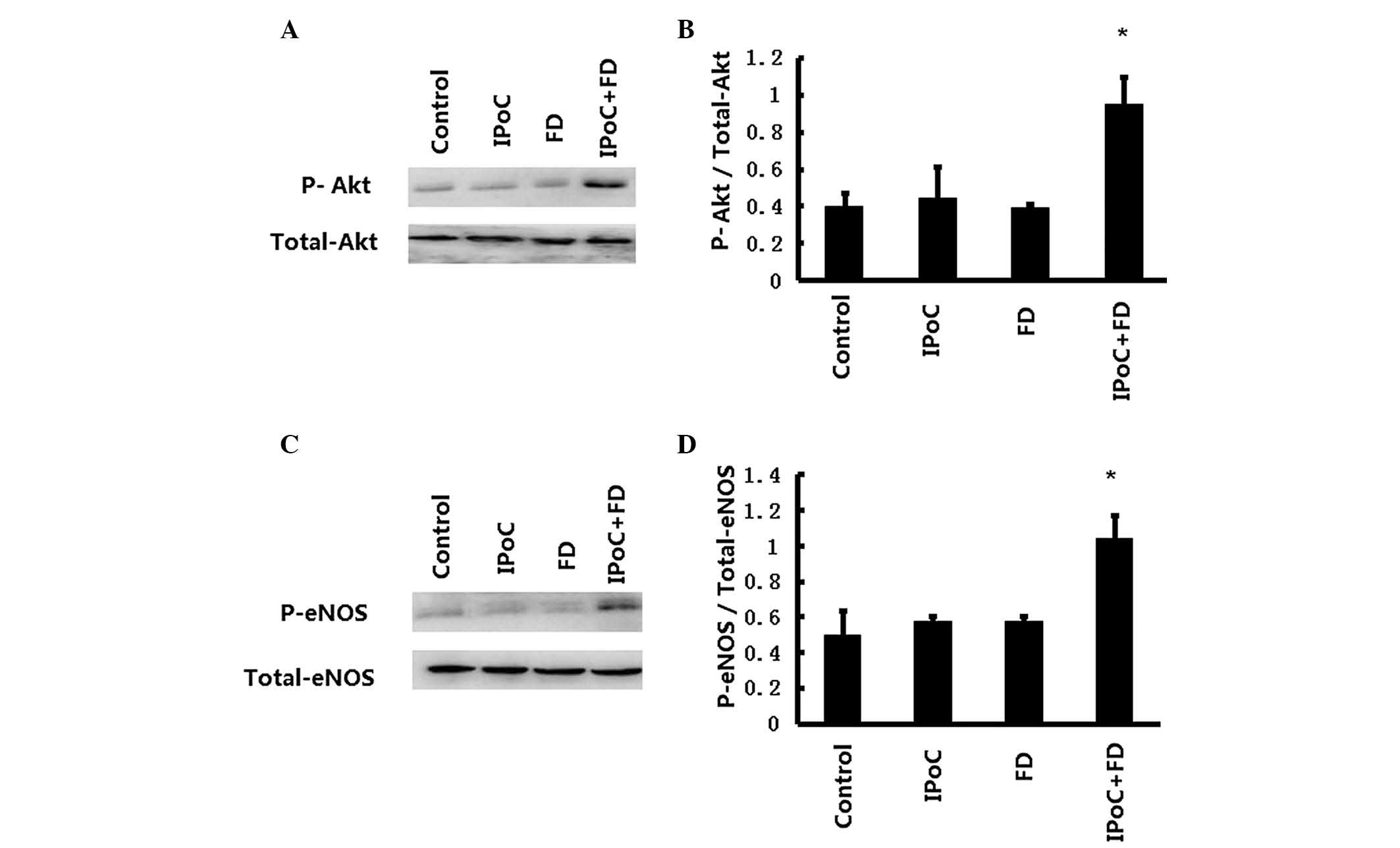
Protein expression of Akt and eNOS
As demonstrated in Fig.
5A and B, the Akt phosphorylation was enhanced in IPoC+FD
group, as compared with the control group (0.951±0.146 vs.
0.402±0.166, P<0.05), but there is no significant change in Akt
phosphorylation in the IPoC and FD groups, as compared with the
control group (0.445±0.170 and 0.390±0.024, respectively, vs.
0.402±0.166, P>0.05). Furthermore, as revealed in Fig. 5C and D, the eNOS phosphorylation
was also enhanced in the IPoC+FD group as compared with the control
group (1.043±0.124 vs. 0.516±0.037, P<0.05), whereas no
significant change in eNOS phosphorylation was demonstrated in the
IPoC and FD groups as compared with the control group (0.577±0.023
and 0.575±0.026, respectively, vs. 0.516±0.037, P>0.05).
Effects of wortmannin and L-NAME on the
cardioprotection of fasudil combined with IPoC treatment
To further assess the role of PI3K/Akt/eNOS pathway
in the restoration of cardioprotection of IPoC in
hypercholesterolemic myocardium by fasudil, the PI3K/Akt specific
inhibitor, wortmannin and eNOS specific inhibitor, L-NAME in
fasudil combined with IPoC treatment group were administered. The
results demonstrated that the cardioprotection of fasudil combined
with IPoC treatment in hypercholesterolemic myocardium was
abrogated by treatment with wortmannin and L-NAME alone, as
evidenced by no significant differences in hemodynamic parameters,
myocardial infarct size, the percentage of cardiomyocyte apoptosis
and the content of NO were demonstrated in IPoC+FD group, as
compared with the control group.
Discussion
The present study demonstrates for the first time,
to the best of our knowledge, that administration of fasudil
shortly prior to ischemia may restore the cardioprotection of IPoC
in the presence of hypercholesterolemia, while it also enhances the
phosphorylation of eNOS and Akt and confers significant increase in
the content of NO. By contrast, the administration of fasudil at
the same dose and IPoC alone do not appear to exert the
cardioprotection in the presence of hypercholesterolemia.
IPoC is a powerful form of protection, but its
effectiveness under pathological conditions is in dispute. A number
of studies have reported that the cardioprotection of IPoC is
limited in the presence of diabetes, hyperlipidemia, uremia and so
on (21,22). Among these disease conditions,
hyperlipidemia, particularly hypercholesterolemia, is regarded as
an independent risk factor in the development of ischemic heart
diseases, including myocardial infarction. In the present study, it
was identified that the cardioprotection of IPoC was blocked by
elevated level of blood hypercholesterolemia, which was induced by
feeding the rats an 8-week hypercholesterolemic diet. The results
of the present study are consistent with the data found in previous
studies (5–8). However, Donato et al
demonstrated that ischemic postconditioning reduces infarct size by
activation of A1 receptors and K+ATP channels in both
normal and hypercholesterolemic rabbits (23). The discrepancies may possibly be
attributed to the animal species, duration of hyperlipidemia diet
and presence/absence of significant coronary sclerosis.
Furthermore, the present study investigated the influence of
hypercholesterolemia on the efficacy of IPoC in the isolated rat
heart, which is independent of the impact of coronary sclerosis.
Therefore, isolated rat heart reperfusion appears to be a more
suitable model to study the direct effect of hypercholesterolemia
on the cardioprotective mechanisms of IPoC.
The mechanisms that
hyperlipidemia/hypercholesterolemia abrogates the cardioprotective
effects of ischemic postconditioning remain unclear. Kupai et
al found experimental hyperlipidemia induced by
cholesterol-enriched diet impaired the cardioprotective effect of
postconditioning by alternating the nitrosative stress signal and
increasing the production of several oxidants, including
peroxynitrite and lipid peroxidation compounds (7). In addition, a long term (3-week) or a
short-term (3-day) statin administration restored the infarct
size-limiting effect of postconditioning in hypercholesterolemic
rat heart, potentially by increasing the expression and activity of
eNOS (9,24).
Previous studies have revealed that Rho-kinase, a
serine/threonine kinase is involved in numerous cardiac
pathological conditions (25). It
is abnormally activated in ischemic myocardium, suggesting that
abnormal activation of Rho-kinase may be correlated with
ischemia-induced myocardial injury (26). In the present study, it was
identified that hypercholesterolemia blocked the cardioprotection
of IPoC, which was accompanied by with the upregulation of the
activity of Rho-kinase, as evidenced by the elevated level of
MYPT-1 phosphorylation, a marker of Rho-kinase activity, in the
IPoC group. The present study further determined that 1 μM fasudil
administration prior to ischemia restored the cardioprotection of
IPoC by decreasing Rho-kinase activity as evidenced by a lower
expression of MYPT-1 phosphorylation in the FD+IPoC group, whereas
administration of fasudil at the same dose and IPoC alone were
ineffective in decreasing the expression of MYPT-1
phosphorylation.
NO is an important messenger in cardiovascular
regulation and also has an important role in protecting the
myocardium against ischemia reperfusion injury (27). It was reported that L-arginine, a
precursor of NO, improved post-ischemic functional recovery and
limited infarct size in the isolated rat heart. Furthermore, the
NOS inhibitor L-NAME eliminated the effect of L-arginine on
ischemia reperfusion injury (28,29).
The main source of cardiac NO is generated through eNOS expressed
by coronary endothelial cells and cardiac myocytes, which is
regulated by PI3K/Akt (30).
Hypercholesterolemia is reported to blunt the infarct size-limiting
effect of IPoC possibly by decreasing cardiac NO content (7). Hypercholesterolemia also decreases NO
bioavailability by downregulating eNOS, in association with an
increased production of oxygen-derived free radicals that may
inactivate NO (31,32). It is reported that fasudil may lead
to the activation of the PI3K/Akt/eNOS signaling pathway and
increased the NO content of myocardium under the condition of
ischemia reperfusion (16). In the
present study, it was demonstrated fasudil restored the
cardioprotection of IPoC in the hypercholesterolemic rat heart by
elevating the phosphorylation of Akt and eNOS, and increasing the
NO content of myocardium. The results suggested that fasudil
restored the cardioprotection of IPoC in hypercholesterolemic rat
heart possibly by upregulating PI3K/Akt/eNOS signal pathway to
increase the synthesis of NO. This hypothesis was further confirmed
as evidenced by the fact that the cardioprotective effects of
fasudil combined with IPoC treatment in the hypercholesterolemic
rat heart, were eliminated by administration of the PI3K specific
inhibitor, wortmannin, and the eNOS specific inhibitor, L-NAME.
Compared with preconditioning which must be applied
prior to an ischemic event, postconditiong has the advantage that
it may be applied following sustained ischemia. It has a notably
more extensive clinical applicability. However, mounting randomized
controlled trials results have demonstrated that ST-elevation
myocardial infarction patients perform IPoC during primary
percutaneous coronary intervention does not reduce myocardial
damage, and it may even aggravate myocardial reperfusion injury
(33,34). The reason for such results may be
that clinical patients tend to have numerous complications,
including diabetes, hyperlipidemia, renal dysfunction etc. which
may affect the effectiveness of IPoC. Therefore, using drugs that
reverse the cardioprotection of IPoC in the state of
hypercholesterolemia are of marked importance. The present study
found that fasudil, a specific Rho-kinase inhibitor which is
approved for human use, restores the protective effect of IPoC in
hyperlipidemic/hypercholesterolemic animal heart. These data may
provide a new drug to restore the cardioprotective effect of IPoC
in patients with other pathological complications.
In conclusion, it was demonstrated that the
inhibition of Rho-kinase by fasudil was able to restore the
cardioprotection of IPoC in the hypercholesterolemic rat heart. The
mechanism underlying this effect appears to involve the
upregulation of PI3K/Akt/eNOS signal pathway and an increase in
myocardial NO content.
Acknowledgements
The present study was supported by Liaoning
Provincial Science and Technology Projects, Liaoning, China (grant
no. 2013021011).
References
|
1
|
Zhao ZQ, Corvera JS, Halkos ME, et al:
Inhibition of myocardial injury by ischemic postconditioning during
reperfusion: comparison with ischemic preconditioning. Am J Physiol
Heart Circ Physiol. 285:H579–H588. 2003.PubMed/NCBI
|
|
2
|
Schwartz LM and Lagranha CJ: Ischemic
postconditioning during reperfusion activates Akt and ERK without
protecting against lethal myocardial ischemia-reperfusion injury in
pigs. Am J Physiol Heart Circ Physiol. 290:H1011–H1018. 2006.
View Article : Google Scholar
|
|
3
|
Gross GJ, Gauthier KM, Moore J, et al:
Evidence for role of epoxyeicosatrienoic acids in mediating
ischemic preconditioning and postconditioning in dog. Am J Physiol
Heart Circ Physiol. 297:H47–H52. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Staat P, Rioufol G, Piot C, et al:
Postconditioning the human heart. Circulation. 112:2143–2148. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Iliodromitis EK, Zoga A, Vrettou A, et al:
The effectiveness of postconditioning and preconditioning on
infarct size in hypercholesterolemic and normal anesthetized
rabbits. Atherosclerosis. 188:356–362. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhao JL, Yang YJ, You SJ, Cui CJ and Gao
RL: Different effects of postconditioning on myocardial no-reflow
in the normal and hypercholesterolemic mini-swines. Microvasc Res.
73:137–142. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kupai K, Csonka C, Fekete V, et al:
Cholesterol diet-induced hyperlipidemia impairs the
cardioprotective effect of postconditioning: role of peroxynitrite.
Am J Physiol Heart Circ Physiol. 297:H1729–H1735. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lauzier B, Delemasure S, Pesant M, et al:
A cholesterol-rich diet improves resistance to ischemic insult in
mouse hearts but suppresses the beneficial effect of
post-conditioning. J Heart Lung Transplant. 28:821–826. 2009.
View Article : Google Scholar
|
|
9
|
Andreadou I, Farmakis D, Prokovas E, et
al: Short-term statin administration in hypercholesterolaemic
rabbits resistant to postconditioning: effects on infarct size,
endothelial nitric oxide synthase, and nitro-oxidative stress.
Cardiovasc Res. 94:501–509. 2012. View Article : Google Scholar
|
|
10
|
Zhang FJ, Ma LL, Wang WN, et al:
Hypercholesterolemia abrogates sevoflurane-induced delayed
preconditioning against myocardial infarct in rats by alteration of
nitric oxide synthase signaling. Shock. 37:485–491. 2012.
View Article : Google Scholar
|
|
11
|
Ma LL, Zhang FJ, Qian LB, et al:
Hypercholesterolemia blocked sevoflurane-induced cardioprotection
against ischemia-reperfusion injury by alteration of the
MG53/RISK/GSK3β signaling. Int J Cardiol. 168:3671–3678.
2013.PubMed/NCBI
|
|
12
|
Fukumoto Y, Mohri M, Inokuchi K, et al:
Anti-ischemic effects of fasudil, a specific Rho-kinase inhibitor,
in patients with stable effort angina. J Cardiovasc Pharmacol.
49:117–121. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu DJ, Xu JZ, Wu YJ, et al: Effects of
fasudil on early atherosclerotic plaque formation and established
lesion progression in apolipoprotein E-knockout mice.
Atherosclerosis. 207:68–73. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ho TJ, Huang CC, Huang CY and Lin WT:
Fasudil, a Rho-kinase inhibitor, protects against excessive
endurance exercise training-induced cardiac hypertrophy, apoptosis
and fibrosis in rats. Eur J Appl Physiol. 112:2943–2955. 2012.
View Article : Google Scholar
|
|
15
|
Wang N, Guan P, Zhang JP, et al: Fasudil
hydrochloride hydrate, a Rho-kinase inhibitor, suppresses
isoproterenol-induced heart failure in rats via JNK and ERK1/2
pathways. J Cell Biochem. 112:1920–1929. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wolfrum S, Dendorfer A, Rikitake Y, et al:
Inhibition of Rho-kinase leads to rapid activation of
phosphatidylinositol 3-kinase/protein kinase Akt and cardiovascular
protection. Arterioscler Thromb Vasc Biol. 24:1842–1847. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li Y, Zhu W, Tao J, et al: Fasudil
protects the heart against ischemia-reperfusion injury by
attenuating endoplasmic reticulum stress and modulating SERCA
activity: the differential role for PI3K/Akt and JAK2/STAT3
signaling pathways. PLoS One. 7:e481152012. View Article : Google Scholar
|
|
18
|
Jiang ZH, Zhang TT and Zhang JF:
Protective effects of fasudil hydrochloride post-conditioning on
acute myocardial ischemia/reperfusion injury in rats. Cardiol J.
20:197–202. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhao H, Wang Y, Wu Y, et al:
Hyperlipidemia does not prevent the cardioprotection by
postconditioning against myocardial ischemia/reperfusion injury and
the involvement of hypoxia inducible factor-1alpha upregulation.
Acta Biochim Biophys Sin (Shanghai). 41:745–753. 2009. View Article : Google Scholar
|
|
20
|
Lv Y, Ren Y, Sun L, Wang S, Wei M and Jia
D: Protective effect of Na(+)/Ca(2+) exchange
blocker KB-R7943 on myocardial ischemia-reperfusion injury in
hypercholesterolemic rats. Cell Biochem Biophys. 66:357–363.
2013.
|
|
21
|
Byrne CJ, McCafferty K, Kieswich J, et al:
Ischemic conditioning protects the uremic heart in a rodent model
of myocardial infarction. Circulation. 125:1256–1265. 2012.
View Article : Google Scholar
|
|
22
|
Ferdinandy P, Schulz R and Baxter GF:
Interaction of cardiovascular risk factors with myocardial
ischemia/reperfusion injury, preconditioning, and postconditioning.
Pharmacol Rev. 59:418–458. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Donato M, D’Annunzio V, Berg G, et al:
Ischemic postconditioning reduces infarct size by activation of A1
receptors and KATP channels in both normal and hypercholesterolemic
rabbits. J Cardiovasc Pharmacol. 49:287–292. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Iliodromitis EK, Andreadou I, Prokovas E,
et al: Simvastatin in contrast to postconditioning reduces infarct
size in hyperlipidemic rabbits: possible role of
oxidative/nitrosative stress attenuation. Basic Res Cardiol.
105:193–203. 2010. View Article : Google Scholar
|
|
25
|
Noma K, Oyama N and Liao JK: Physiological
role of ROCKs in the cardiovascular system. Am J Physiol Cell
Physiol. 290:C661–C668. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hamid SA, Bower HS and Baxter GF: Rho
kinase activation plays a major role as a mediator of irreversible
injury in reperfused myocardium. Am J Physiol Heart Circ Physiol.
292:H2598–H2606. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jugdutt BI: Nitric oxide and
cardioprotection during ischemia-reperfusion. Heart Fail Rev.
7:391–405. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Suematsu Y, Ohtsuka T, Hirata Y, et al:
L-Arginine given after ischaemic preconditioning can enhance
cardioprotection in isolated rat hearts. Eur J Cardiothorac Surg.
19:873–879. 2001. View Article : Google Scholar
|
|
29
|
Izhar U, Schwalb H, Borman JB and Merin G:
Cardioprotective effect of L-arginine in myocardial ischemia and
reperfusion in an isolated working rat heart model. J Cardiovasc
Surg (Torino). 39:321–329. 1998.PubMed/NCBI
|
|
30
|
Dimmeler S, Fleming I, Fisslthaler B, et
al: Activation of nitric oxide synthase in endothelial cells by
Akt-dependent phosphorylation. Nature. 399:601–605. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dauber IM, Lesnefsky EJ, VanBenthuysen RM,
Weil JV and Horwitz ID: Reactive oxygen metabolite scanvengers
decrease functional coronary microvascular injury due to ischemia
reperfusion. Am J Physiol. 260:H42–H49. 1991.
|
|
32
|
Wilson SH, Simari RD, Best PJ, et al:
Simvastatin preserves coronary endothelial function in
hypercholesterolemia in the absence of lipid lowering. Arterioscler
Thromb Vasc Biol. 21:122–128. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Freixa X, Bellera N, Ortiz-Pérez JT, et
al: Ischaemic postconditioning revisited: lack of effects on
infarct size following primary percutaneous coronary intervention.
Eur Heart J. 33:103–112. 2012. View Article : Google Scholar
|
|
34
|
Tarantini G, Favaretto E, Marra MP, et al:
Postconditioning during coronary angioplasty in acute myocardial
infarction: the POST-AMI trial. Int J Cardiol. 162:33–38. 2012.
View Article : Google Scholar : PubMed/NCBI
|