Introduction
Primary hepatocellular carcinoma (HCC) is a common
malignant tumor and a significant public health concern worldwide
(1). Despite the availability of
numerous treatment options for patients with HCC, a high rate of
recurrence and metastasis results in a low five-year survival rate
for this fatal disease (2). There
is therefore an urgent requirement to advance our understanding of
the mechanisms underlying HCC recurrence and metastasis in order to
improve the efficacy of the current treatment strategies. HCC is
similar to numerous tumors, has a rich blood supply. HCC relies on
the formation of blood vessels for growth and metastasis (3). Although HCC is a highly angiogenic
cancer, it is characterized by hypoxia due to its rapid growth rate
and numerous hypovascular areas (4). HCC cells are frequently under hypoxic
conditions and recent evidence suggests that hypoxia is crucially
involved in tumor progression and angiogenesis. The formation of
new vessels in the tumor is controlled simultaneously by pro- and
anti-angiogenic factors. The most prominent of several
pro-angiogenic factors is vascular endothelial growth factor (VEGF)
(5). The upregulation of VEGF
expression in tumor tissues has been reported to be associated with
poor prognosis in several cancers, including HCC (6).
Previous studies have provided strong evidence that
the interaction between tumor cells and their microenvironment,
consisting of stromal cells and the extracellular matrix (ECM),
contributes to cancer progression (7,8). In
the majority of tumors, an abnormal network of stromal cells,
growth factors, cytokines and chemokines is critical for the
induction of angiogenesis (9,10).
The cancer hypoxic microenvironment creates a suitable condition
for neo-vascularization by enhancing the expression of
pro-angiogenic factors and reducing that of anti-angiogenic factors
(3,11). In HCC, VEGF has been reported to be
produced by both HCC and stromal cells (10).
The majority of patients with HCC have a history of
chronic liver disease, and the presence of liver cirrhosis is
closely associated with the development of HCC (12). Activation of hepatic stellate cells
(HSCs) is a fundamental step in the development of liver fibrosis
and ultimately cirrhosis (13). In
addition to these actions, previous studies have indicated that
activated HSCs infiltrate the liver tumor stroma and become one of
the most prominent stromal cell types in the liver tumor
environment (14,15). Clinical evidence has revealed that
the presence of peritumoral HSCs correlates with the recurrence of
HCC (16). Although some data have
suggested an important contribution of HSCs to HCC proliferation
and metastasis, the exact molecular interactions that occur between
these two cell types are unknown. It has been reported that
activated HSCs can interact with HCC cells in a paracrine manner
(17,18). In the process of liver fibrosis and
portal hypertension, activated HSCs have been reported to
synthesize pro-angiogenic factors, including VEGF, angiopoietin-1
and several metalloproteinases (19). In experimentally induced liver
metastases, activated HSCs have been shown to be pro-angiogenic
during the progression of liver cancer (20). These studies suggest that HSCs may
contribute to the angiogenic requirement of HCC proliferation and
metastasis. The molecular mechanisms of this process remain
unclear.
Platelet-derived growth factor (PDGF) is a potent
mitogen for mesenchymal cells that is synthesized, stored and
released by numerous cell types, including tumor cells (21). PDGF is a dimeric glycoprotein that
is composed of two A chains (PDGF-AA), two B chains (PDGF-BB) or a
combination of the two (PDGF-AB). Under the hypoxic tumor
microenvironment, accumulated evidences indicate that expression of
PDGF-BB is elevated in tumor cells and potently stimulates several
biological actions in HSCs (22).
In colorectal liver metastasis, tumor cell-derived PDGF-BB promotes
tumor growth through a growth-promoting effect on HSCs (23). PDGF-BB also affects the angiogenic
properties of HSCs during liver fibrosis (24). In liver cancer, however, the
function of PDGF-BB in HSCs has not been well characterized.
From these observations, the present study has
speculated that HCC-derived factors may influence activated HSCs to
regulate angiogenesis under the hypoxic HCC microenvironment. The
present study aimed to explore this mechanism through in
vitro analysis of the paracrine effects of secreted,
HCC-derived PDGF-BB on the proliferation, migration and
pro-angiogenic gene expression of HSCs in hypoxia.
Materials and methods
Cell culture
The HepG2 liver cancer cell line was purchased from
the Animal Center Laboratory of Sun Yat-sen University (Guangzhou,
China). The LX-2 activated hepatic stellate cell line was obtained
from the American Type Culture Collection (ATCC, Manassas, VA, US).
All cells were routinely cultured in high-glucose Dulbecco’s
Modified Eagle’s Medium (DMEM) (Gibco-BRL, Grand Island, NY, US)
supplemented with 10% fetal bovine serum (FBS) (HyClone
Laboratories, South Logan, UT, USA) at 37°C with 5% CO2
and 95% air in a humidified incubator (25). The hypoxic stimulation of HepG2
cells was achieved by exposing the cells to 1% O2, 5%
CO2 and 94% N2 in an incubator as previously
described (26). An indirect
co-culture of LX-2 and HepG2 cells was assembled using Transwell
membranes (24 mm diameter, 0.4 μm pore size; Corning Costar, Acton,
MA, USA). A total of ~1×104 LX-2 cells were seeded in
the lower chamber, and 1×103 HepG2 cells were seeded on
the membrane insert. The co-cultures were maintained for 72 h.
Collection of conditioned medium
(CM)
To prepare the CM, the cells were washed twice with
serum-free DMEM one day after being seeded into T75 flasks
(1×106 cells). The cells were incubated with serum-free
DMEM under normoxic conditions for 24 h or under hypoxic conditions
for 12 or 24 h. Simultaneously, serum-free DMEM was placed in
cell-free culture flasks under the same conditions to serve as a
control. For inhibition experiments, CM from HepG2 cells was
preincubated with a PDGF-BB neutralizing antibody at a
concentration of 5 μg/ml as previously described (27).
Expression constructs and
transfection
The expression construct for full-length human
PDGF-BB was generated by cloning a polymerase chain
reaction-amplified PDGF-BB cDNA fragment into the
pCDH-CMV-MCS-EF1-copGFP lentiviral vector, which allowed for stable
transfection. Virus packaging was performed in HEK293T cells
following cotransfection of the recombinant lentiviral expression
and packaging plasmids (pGag/Pol, pRev and pVSV-G) using
Lipofectamine™ 2000 (Invitrogen Life Technologies, Carlsbad, CA,
USA). Target cells were infected with the harvested virus for 12 h,
and the original medium was replaced with fresh medium. The cells
expressing PDGF-EGFP were sorted by flow cytometry. The lentiviral
transduction efficiency was analyzed by western blotting.
MTT assay
The proliferation rate of LX-2 cells was measured
using an MTT assay. Briefly, LX-2 cells were seeded in 96-well
plates at a density of 2×104 cells/well. After 24 h, CM
was added to the wells, and the plates were incubated at 37°C for
an additional 24 h. MTT (50 μl of 5 mg/ml) was added to the
culture, which was then further incubated for 4 h at 37°C. The
optical density was measured at 490 nm using a plate reader. The
absorbance values directly correlated with the number of
proliferating cells in the culture.
Migration assay
To determine whether the HepG2 cells attracted the
LX-2 cells, a migration assay was performed using Transwell
chambers containing polycarbonate filters with 8 μm pores (Corning
Costar). The lower compartment contained CM. The LX-2 cells were
harvested and resuspended in DMEM without FBS, at a density of
5×104 cells/ml and placed in the upper chamber.
Following incubation at 37°C for 24 h, the filters were collected,
and the cells that adhered to the lower surface were fixed, stained
for 1 h with crystal violet (Sigma, St. Louis, MO, USA) in 2%
ethanol and then rinsed in water. The number of cells that migrated
across the filters was counted using a Nikon TS100 microscope
(Nikon Corporation, Tokyo, Japan) in four high-power fields per
insert and average values were calculated. The experiments were
performed in triplicate with consistent results.
Western blot analysis
Cells were collected in phosphate-buffered saline
(PBS) and lysed on ice for 30 min in radioimmunoprecipitation assay
lysis buffer. The protein content in the cell lysates were
quantitated using the Bradford reagent. Equal amounts of protein
lysate (30 μg) were resolved by 10% SDS-PAGE and
electrophoretically transferred onto polyvinylidene fluoride
membranes. The membranes were then blocked in 5% nonfat dried milk
for 1 h. The blots were probed with the indicated primary
antibodies (PDGF-BB, PDGFR-β or VEGF-A) overnight at 4°C and then
with the appropriate horseradish peroxidase-conjugated secondary
antibody (1:5,000) for 1 h at room temperature. The blots were
developed by enhanced chemiluminescence (Santa Cruz, CA, USA) and
exposed to X-ray film. GAPDH was used as a loading control.
ELISA
PDGF-BB is a secreted protein, therefore, the
concentration of PDGF-BB in the culture medium was measured to
estimate the expression level of PDGF-BB in HepG2 cells. To compare
the expression level of PDGF-BB under normoxia and hypoxia, the
amount of PDGF-BB in the HepG2 supernatant was analyzed using a
commercial ELISA kit (eBioscience, San Diego, CA, USA) according to
the manufacturer’s instructions.
Statistical analysis
All data are expressed as the mean ± standard error
of the mean, from at least three independent experiments.
Statistical analyses were performed using the SPSS statistical
software for Microsoft Windows, version 13.0 (SPSS, Inc., Chicago,
IL, USA). A two-tailed paired Student’s t-test and analysis of
variance was used to determine significance between the test and
the control conditions. A P<0.05 was considered to indicated a
statistically significant difference.
Results
Effects of hypoxia on PDGF-BB expression
in HepG2 cells
The basal expression of PDGF-BB in HepG2 cells and
the change in expression following exposure to hypoxia was
analyzed. HepG2 cells were cultured in an atmosphere containing
either 21% oxygen (normoxia) or 1% oxygen (hypoxia) for 12 or 24 h.
At the end of the incubation, PDGF-BB expression was assessed by
western blotting. As shown in Fig.
1A, there was little PDGF-BB expression in HepG2 cells cultured
under normoxia. As compared with normoxia, exposure to hypoxia
increased the protein expression of PDGF-BB, and this increase was
more robust at 24 h (Fig. 1A). An
ELISA was used to detect the secretion of PDGF-BB by HepG2 cells
and the PDGF-BB protein concentrations under normoxic and hypoxic
conditions were compared. A low concentration of PDGF-BB was
detected in the supernatant of HepG2 cells cultured in normoxia,
and this concentration significantly increased after exposure to
hypoxia (Fig. 1B). These results
indicated that PDGF-BB is expressed in HCC cells and is secreted to
act in a paracrine manner. Furthermore, the data indicated that
hypoxia increased the production and secretion of PDGF-BB by HCC
cells.
PDGFR-β expression in LX-2 cells
increases when co-cultured with HepG2 cells
The activated LX-2 HSC cell line was analyzed for
the presence of PDGFR-β by western blotting. LX-2 cells were
co-cultured with HepG2 cells for 72 h, and the level of PDGF
receptor-β (PDGFR-β) expression in LX-2 cells grown in monoculture
was compared with that of LX-2 cells grown in co-culture. The
results confirmed that PDGFR-β is expressed in LX-2 cells grown in
monoculture. When co-cultured with HepG2 cells, the protein
expression of PDGFR-β in LX-2 cells increased (Fig. 2), indicating that HCC cells induced
PDGFR-β expression in activated HSCs. These results suggest that
HCC cells affect the biological function of activated HSCs via the
paracrine action of PDGF-BB.
HepG2 cells increased the proliferation
and migration of LX-2 cells
To examine the effects of HepG2 cells on LX-2 cell
proliferation, LX-2 cells were incubated with CM from HepG2 cells
cultured under normoxia or hypoxia. An MTT assay was used to
measure the proliferation rate of the LX-2 cells. Compared with
control medium, HepG2-CM promoted LX-2 cell growth in vitro.
HepG2-CM that was collected after culture in hypoxia had a greater
effect on LX-2 cell proliferation at various time points (Fig. 3A).
A cell migration assay was used to investigate
whether HepG2 cells attracted LX-2 cells. In this experiment,
24-well Transwell plates with 8 μm pores were used. A total of
~1×105 LX-2 cells were suspended in serum-free medium
and placed in the insert. Control or CM that was freshly obtained
from HepG2 cells under normoxia or hypoxia was added to the lower
chamber. As compared with the control medium, HepG2-CM
significantly stimulated the migration of LX-2 cells. HepG2-CM
collected from hypoxic culture conditions had a greater effect on
LX-2 cell migration than that collected from normoxic culture
conditions (Fig. 3B).
HepG2-derived PDGF-BB increases LX-2 cell
proliferation and migration
Previous studies have suggested that HepG2 cells
increase the proliferation and migration of LX-2 cells and that
this effect is greater when HepG2 cells are exposed to hypoxia. To
investigate whether PDGF-BB was causative of these observations, an
HepG2 cell line that stably overexpressed PDGF-BB (HepG2-PDGF) was
construced. In addition, a PDGF-BB neutralizing antibody was used
to inhibit PDGF-BB protein in the HepG2 supernatant, as a negative
control. The MTT and cell migration assays were repeated to
determine whether HepG2-derived PDGF-BB affects the proliferation
and migration of LX-2 cells. As in the previous experiments, the
hypoxic 24 h group was used as the control and the proliferation
rate was analyzed at 48 h. The highest PDGF-BB concentration in the
HepG2-PDGF supernatant among all of the groups was confirmed by
ELISA (data not shown). The CM from the HepG2-PDGF cells
significantly increased LX-2 cell proliferation and migration. The
effect of the HepG2-CM on LX-2 cell proliferation and migration was
mitigated by the presence of the PDGF-BB neutralizing antibody
(Fig. 4A and B). Together, these
data suggest that HCC cells stimulate the proliferation of LX-2
cells in a PDGF-BB-dependent paracrine manner.
Effect of HepG2 cells on VEGF-A
expression in LX-2 cells
Recent studies have suggested that activated HSCs
express pro-angiogenic genes that are involved in intrahepatic
angiogenesis in chronic hepatic disease. It was investigated
whether HepG2 cells could stimulate LX-2 cells to express VEGF-A,
the most prominent protein involved in angiogenesis. LX-2 cells
were incubated for 48 h with CM collected from HepG2 cells cultured
under normoxia or hypoxia. The VEGF-A expression in LX-2 cells was
determined by western blotting. As compared with the control group,
HepG2-CM stimulated the expression of VEGF-A in the LX-2 cells. CM
collected from HepG2 cells under hypoxia robustly increased VEGF-A
expression in the LX-2 cells (Fig.
5). These results suggest that HCC cells stimulate the
pro-angiogenic activity of activated HSCs in a paracrine
manner.
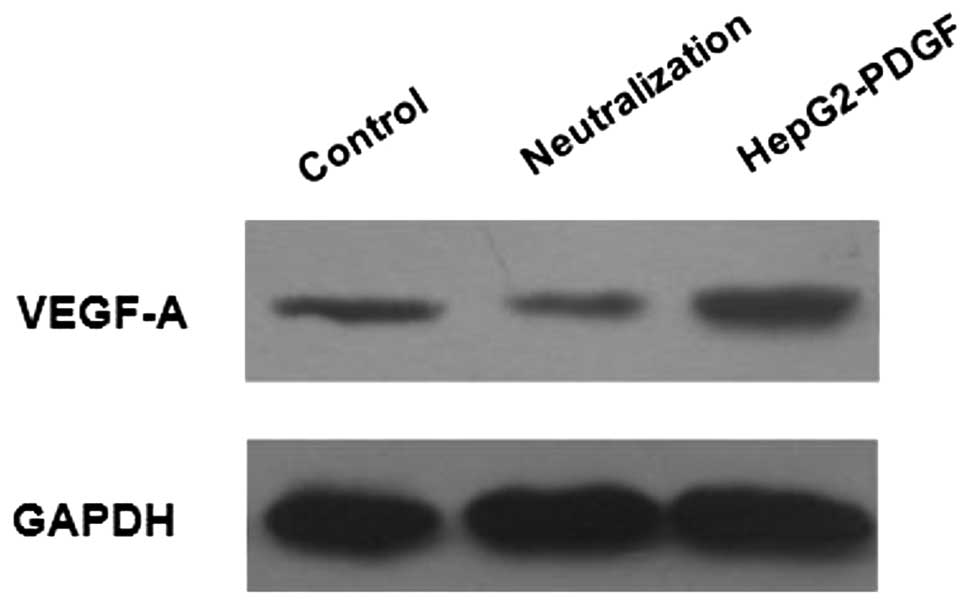
HepG2-derived PDGF-BB increases VEGF-A
expression in LX-2 cells
It was aimed to determine whether PDGF-BB, secreted
by HepG2 cells, was responsible for the upregulation of VEGF-A
expression in LX-2 cells. LX-2 cells were incubated for 48 h with
CM collected from HepG2-PDGF cells or with HepG2-CM pre-treated
with the PDGF-BB neutralizing antibody. The HepG2-CM from cells
exposed to hypoxia for 24 h was used as a control. Western blot
analysis demonstrated that CM from HepG2-PDGF cells significantly
increased the expression of VEGF-A in LX-2 cells as compared with
the control. In the presence of the PDGF-BB neutralizing antibody,
the HepG2-CM-mediated effect on VEGF-A expression in LX-2 cells was
diminished (Fig. 6). These results
indicated that the PDGF-BB secreted by HCC cells stimulated the
pro-angiogenic properties of HSCs.
Discussion
The present study has demonstrated that hypoxia
induces HCC cells to secrete PDGF-BB. PDGF-BB derived from HCC
cells can mediate the stimulatory and chemoattractant properties of
human HCC cells on HSCs and enhance VEGF-A expression in HSCs. VEGF
has been reported to be one of the strongest pro-angiogenic factors
for tumor angiogenesis (5,28). These data expand on previous
observations that HSCs accumulate in the microenvironment of
hepatic tumors, are associated with tumor angiogenesis (15) and promote tumor proliferation and
metastasis. The present study confirmed that the PDGF-BB secreted
by HCC cells functions in this process, but additional studies are
required to clarify the mechanism.
Tumor progression requires the formation of new
blood vessels (4). The
interactions between tumor and stromal cells are important for the
abnormal angiogenesis that occurs in tumors, and these interactions
are currently under investigation (10). In the tumor microenvironment,
various stromal cells are recruited to and localize within the
tumor stroma. The stromal cells and their secreted factors
constitute an appropriate microenvironment for tumor angiogenesis
(29). Activated HSCs are an
important type of stromal cell in HCC. Numerous studies have
reported that HSCs accumulate within HCC nodules in patients with
liver cirrhosis (13,30). In the present study, CM from HCC
cells significantly increased the proliferation and migration of
HSCs in vitro. This indicates that HCC cells act in a
paracrine manner to attract HSCs and encourage their accumulation
within the HCC stroma. Activated HSCs secrete numerous proteins
under pathological conditions. These cells have pro-angiogenic
properties and interact with endothelial cells that express
pro-angiogenic proteins, including VEGF and angiopoietin (19). It was additionally shown that HCC
cells increased the expression of VEGF-A in HSCs in vitro,
indicating that HSCs in the HCC stroma may participate in tumor
angiogenesis.
PDGF-BB is one of five PDGF isoforms, and is a
potent mitogen for mesenchymal cells, such as smooth muscle and
glial cells (21). Following
binding of PDGF-BB to PDGFR, downstream signaling is initiated that
leads to cell cycle regulation, migration and differential gene
expression (21). PDGFR-β is the
highest affinity receptor for PDGF-BB (31) and in the process of liver fibrosis,
PDGFR-β has been reported to be a key marker of HSC activation
(30). The present study confirmed
that PDGF-BB was expressed in HepG2 cells. The expression of
PDGFR-β in LX-2 cells was confirmed, and it was demonstrated that
this expression increased when the HSCs were co-cultured together
with HCC cells. These results indicate that HCC cells interact with
HSCs through the paracrine functions of PDGF-BB. This study
demonstrated that PDGF-BB, derived from HCC cells, drives the
proliferation, migration and pro-angiogenic protein expression of
HSCs. It was speculated that HCC cells secrete PDGF-BB to promote
the migration and accumulation of HSCs within the tumor stroma. The
induced proliferation and upregulation of VEGF-A expression in HSCs
by HCC cell-derived PDGF-BB may create a pro-angiogenic
microenvironment that encourages HCC angiogenesis. This process is
beneficial for the proliferation and metastasis of HCC. The data
from the present study indirectly supports this hypothesis. Since
only in vitro data were obtained in this study, additional
studies using experimental models of HCC and samples from patients
with advanced liver cirrhosis and HCC should be performed to
confirm this hypothesis.
Hypoxia often exists during tumor progression due to
the rapid tumor growth rate and relative lack of a blood supply
(32). To study the paracrine
interactions between HSCs and HCC cells, the effects of hypoxia on
the paracrine signaling of HCC cells was examined. The effects of
hypoxia on PDGF-BB expression in HCC cells was studied by culturing
HCC cells under hypoxic conditions. The concentration of oxygen in
the culture atmosphere was 1% as compared with 20% in normoxia.
This system has been widely used to explore the effect of hypoxia
on cells (26). In the present
study, evidence was shown supporting that PDGF-BB expression in HCC
cells increase with time in hypoxia. Hypoxia is an important
regulator of angiogenesis in HCC, but the mechanism has not been
fully elucidated. It was found that the expression of VEGF-A in
HSCs increased in response to the hypoxia-induced secretion of
PDGF-BB by HCC cells. The enhanced VEGF-A expression in HSCs may
have an important function in HCC angiogenesis. Future in
vivo studies should investigate whether inhibiting PDGF-BB
expression in HCC cells, or targeting PDGFR-β in HSCs, will affect
HCC angiogenesis and reduce the growth and metastasis of this
aggressive tumor.
In conclusion, the results of the present study have
demonstrated that PDGF-BB secreted by HCC cells promotes the
proliferation, migration and VEGF-A expression of HSCs and that
hypoxia can affect the expression of PDGF-BB in HCC cells. The data
indicate that there is cross-talk between HCC cells and HSCs and
the enhanced expression of VEGF-A may affect HCC angiogenesis.
PDGF-BB is an important mediator of this interaction. Although a
direct interaction between activated HSCs and HCC cells has not
been identified, the shared roles of activated HSCs and HCC cells
in tumor progression suggest a new therapeutic approach of
simultaneously targeting tumor cells and activated HSCs to
effectively prevent the development of HCC.
Acknowledgements
This study was supported by the National Nature
Science Foundation of China (grant nos. 81272642 and 81000177) and
the Young Teacher Training Program of Sun Yat-sen University (grant
no. 11ykpy40).
References
|
1
|
Tang ZY, Ye SL, Liu YK, et al: A decade’s
studies on metastasis of hepatocellular carcinoma. J Cancer Res
Clin Oncol. 130:187–196. 2004.
|
|
2
|
Schwartz M, Roayaie S and Konstadoulakis
M: Strategies for the management of hepatocellular carcinoma. Nat
Clin Pract Oncol. 4:424–432. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Keating GM and Santoro A: Sorafenib: a
review of its use in advanced hepatocellular carcinoma. Drugs.
69:223–240. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Coulon S, Heindryckx F, Geerts A, et al:
Angiogenesis in chronic liver disease and its complications. Liver
Int. 31:146–162. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kraizer Y, Mawasi N, Seagal J, et al:
Vascular endothelial growth factor and angiopoietin in liver
regeneration. Biochem Biophys Res Commun. 287:209–215. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schoenleber SJ, Kurtz DM and Talwalkar JA:
Prognostic role of vascular endothelial growth factor in
hepatocellular carcinoma: systematic review and meta-analysis. Br J
Cancer. 100:1385–1392. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Galiè M, Sorrentino C, Montani M, et al:
Mammary carcinoma provides highly tumourigenic and invasive
reactive stromal cells. Carcinogenesis. 26:1868–1878.
2005.PubMed/NCBI
|
|
8
|
Hernandez-Gea V, Toffanin S, Friedman SL
and Llovet JM: Role of the microenvironment in the pathogenesis and
treatment of hepatocellular carcinoma. Gastroenterology.
144:512–527. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Budhu A, Forgues M, Ye QH, et al:
Prediction of venous metastases, recurrence, and prognosis in
hepatocellular carcinoma based on a unique immune response
signature of the liver microenvironment. Cancer Cell. 10:99–111.
2006. View Article : Google Scholar
|
|
10
|
Ribatti D and Vacca A: The role of
microenvironment in tumor angiogenesis. Genes Nutr. 3:29–34. 2008.
View Article : Google Scholar
|
|
11
|
Zhu AX, Duda DG, Sahani DV, et al: HCC and
angiogenesis: possible targets and future directions. Nat Rev Clin
Oncol. 8:292–301. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Guo J and Friedman SL: Hepatic
fibrogenesis. Semin Liver Dis. 27:413–426. 2007. View Article : Google Scholar
|
|
13
|
Friedman SL: Mechanisms of hepatic
fibrogenesis. Gastroenterology. 134:1655–1669. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Enzan H, Himeno H, Iwanmura S, et al:
Alpha-smooth muscle actin-positive perisinusoidal stromal cells in
human hepatocellular carcinoma. Hepatology. 19:895–903. 1994.
|
|
15
|
Thabut D and Shah V: Intrahepatic
angiogenesis and sinusoidal remodeling in chronic liver disease:
new targets for the treatment of portal hypertension? J Hepatol.
53:976–980. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liao R, Sun TW, Yi Y, et al: Expression of
TREM-1 in hepatic stellate cells and prognostic value in hepatitis
B-related hepatocellular carcinoma. Cancer Sci. 103:984–992. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Amann T, Bataille F, Spruss T, et al:
Activatd hepatic stellate cells promote tumorigenicity of
hepatocellular carcinoma. Cancer Sci. 100:646–653. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Faouzi S, Lepreux S, Bedin C, et al:
Activation of cultured rat hepatic stellate cells by tumoral
hepatocytes. Lab Invest. 79:485–493. 1999.PubMed/NCBI
|
|
19
|
Olaso E, Salado C, Egilegor E, et al:
Proangiogenic role of tumor-activated hepatic stellate cells in
experimental melanoma metastasis. Hepatology. 37:674–685. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Heldin CH: Structural and functional
studies on platelet-derived growth factor. EMBO J. 12:4251–4259.
1992.PubMed/NCBI
|
|
21
|
Watanabe A, Sohail MA, Gomes DA, et al:
Inflammasome-mediated regulation of hepatic stellate cells. Am J
Physiol Gatrointest Liver Physiol. 296:G1248–G1257. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bandapalli OR, Macher-Goeppinger S,
Schirmacher P, et al: Paracrine signalling in colorectal liver
metastases involving tumor cell-derived PDGF-C and hepatic stellate
cell-derived PAK-2. Clin Exp Metastasis. 29:409–417. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Aleffi S, Navari N, Delogu W, et al:
Mammalian target of rapamycin mediates the angiogenic effects of
leptin in human hepatic stellate cells. Am J Physiol Gastrointest
Liver Physiol. 301:G210–G219. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jia YL, Shi L, Zhou JN, et al: Epimorphin
promotes human hepatocellular carcinoma invasion and metastasis
through activation of focal adhesion kinase/extracellular
signal-regulated kinase/matrix metalloproteinase-9 axis.
Hepatology. 54:1808–1818. 2011. View Article : Google Scholar
|
|
25
|
Gleadle JM, Ebert BL, Firth JD, et al:
Regulation of angiogenic growth factor expression by hypoxia,
transition metals, and chelating agents. Am J Physiol.
268:C1362–C1368. 1995.PubMed/NCBI
|
|
26
|
Fingas CD, Bronk SF, Werneburg NW, et al:
Myofibroblast-derived PDGF-BB promotes Hedgehog survival signaling
in cholangiocarcinoma cells. Hepatology. 54:2076–2088. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sharma PS, Sharma R and Tyagi T:
VEGF/VEGFR pathway inhibitors as anti-angiogenic agents: present
and future. Curr Cancer Drug Targets. 11:624–653. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhao W, Zhang L, Yin Z, et al: Activated
hepatic stellate cells promote hepatocellular carcinoma development
in immunocompetent mice. Int J Cancer. 129:2651–2661. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liao R, Wu H, Yi Y, et al: Clinical
significance and gene expression study of human hepatic stellate
cells in HBV related-hepatocellular carcinoma. J Exp Clin Cancer
Res. 32:22–32. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bowen-Pope DF and Raines EW: History of
discovery: platelet-derived growth factor. Arterioscler Thromb Vasc
Biol. 11:2397–2401. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lee UE and Freidman SL: Mechanisms of
hepatic fibrogenesis. Best Pract Res Clin Gastroenterol.
25:195–206. 2011. View Article : Google Scholar
|
|
32
|
Villanueva A and Llovet JM: Targeted
therapies for hepatocellular carcinoma. Gastroenterology.
140:1410–1426. 2011. View Article : Google Scholar : PubMed/NCBI
|