Introduction
Melanoma is the fifth most frequently diagnosed type
of malignancy in males and the sixth in females in the USA
(1). Furthermore, its high rate of
invasiveness and dissemination makes surgery an unlikely option,
with the exception of rare cases (2). Despite the development of new
modalities of therapy, the outcome for patients with advanced
melanoma is extremely poor (3).
Current standard treatment includes the single-agent dacarbazine
which improves clinical response but not the median survival
duration (4,5).
Future improvements in melanoma treatment are likely
to arise from novel agents which target molecular pathways that
regulate tumor cell growth and survival. In accordance with present
research development, the mitogen-activated protein kinases (MAPKs)
pathway is an attractive target for therapeutic intervention in
melanoma (6). MAPKs have an
important role in the regulation of numerous cellular processes,
including cell growth and proliferation, differentiation, and
apoptosis. MAPKs consist of extracellular signal-related kinases
(ERKs), c-Jun NH2-terminal kinases (JNKs) and p38 MAPKs
(7). Previous studies have
indicated that the activation of p38 MAPK is involved in cell
growth arrest and apoptosis via the generation of reactive oxygen
species (ROS) (8,9). It has also been reported that p38
activation leads to accumulation of p53, a major tumor suppressor
protein (10). p53-dependent cell
cycle arrest is mainly mediated by transcriptional activation of
p21 (11).
Previous studies indicate that ROS generation, by
which a number of anti-cancer agents act, is in part responsible
for the cytotoxic efficacy in a numerous types of tumor cell
(12,13). Icariside II (IS) is a metabolite of
icariin, which is derived from Herba Epimedii. IS is a novel
anticancer drug that induces apoptosis in tumor cell lines
(14–16). In the present study, the
antiproliferative effects of IS on A375 human melanoma cells in
vitro are evaluated and the possible mechanism through the
ROS-p38-p53 signaling pathway is demonstrated.
Materials and methods
Reagents and cell culture
Icariside II (>98% pure) (Fig. 1A) was isolated by the enzymatic
hydrolysis of icariin (Shanghai Ronghe Pharmaceutical Company,
Shanghai, China), as previously described (17). The A375 human melanoma cells were
purchased from American Type Culture Collection (Manassas, VA, USA)
and maintained in Dulbecco’s modified Eagle’s medium (DMEM;
Invitrogen, Carlsbad, CA, USA) containing 4 mM L-glutamine, 3.7 g/l
sodium bicarbonate, 4.5 g/l glucose and 10% fetal bovine serum
(FBS; Invitrogen). Cells were maintained in a 5% CO2
humidified incubator at 37°C. WST-8 was obtained from Dojindo
(Mashikimachi, Japan), propidium iodide (PI) and RNaseA were
supplied by Beyotime Institute of Biotechnology (Haimen, China).
Rabbit monoclonal (P)-p38, mouse monoclonal P-p53, mouse monoclonal
p21, rabbit polyclonal P-cyclin-dependent kinase 1 (P-CDK1), rabbit
monoclonal cyclin-dependent kinase 2 (CDK2), mouse polyclonal
cyclin E, rabbit monoclonal cyclin B1, rabbit monoclonal cleaved
poly (ADP-ribose) polymerase (PARP) and mouse monoclonal β-actin
antibodies were obtained from Cell Signaling Technology, Inc.
(Beverly, MA, USA). N-acetyl-L-cysteine (NAC), SB203580 and
pifithrin-α were supplied by Sigma-Aldrich (St. Louis, MO, USA).
Horse radish peroxidase-conjugated secondary anti-mouse IgG and
anti-rabbit IgG antibodies were provided by Cell Signaling
Technology, Inc.
Cell viability assays
IS dissolved in dimethylsulfoxide (DMSO) was used
for the treatment of cells. The final concentration of DMSO used
was <0.1% (v/v). Cell viability was measured using the WST-8
assay from Dojindo following the optimized manufacturer’s
instructions. The A375 cells were seeded at a density of 3,000
cells/well in 96-well culture plates in DMEM and incubated in a
humidified incubator at 37°C overnight. The cells were pretreated
with or without NAC (2 mM), SB203580 (5 μM), or pifithrin-α (5 μM)
for 1 h. Then the cells were treated with different concentrations
of IS (0, 25, 50 or 100 μM). After 24 h of post-treatment
incubation, 10 μl WST-8 was added to each well for 1 h.
Subsequently the optical density (OD) was measured at 450 nm. The
percentage of viable cells was determined by the following formula:
Ratio =
[(ODIS-ODblank)/(ODcontrol-ODblank)]
× 100. The cell viability data are averages of three independent
experiments each containing six replicates.
Cell proliferation assays
The A375 cells were seeded at a density of
2×105 cells/well in 6-well culture plates in DMEM and
incubated in a humidified incubator at 37°C for 24 h prior to
treatment with different concentrations of IS (0, 25, 50 or 100
μM). After 24 h of post-treatment incubation, the cells were
harvested and resuspended in 1 ml DMEM. Following resuspension, 100
μl cells were added to a CASY cup containing 10 ml CASY ton (Roche,
Mannheim, Germany), an electrolyte/buffer. The detection of living
and total cell numbers was determined by the Casy Cell Counter and
Analyzer system (Roche) (18,19).
Cell cycle and cell death analysis
For cell cycle analysis, A375 cells were seeded at a
density of 2×105 cells/well in 6-well culture plates in
DMEM and incubated in a humidified incubator at 37°C for 24 h. Then
the cells were starved with fetal bovine serum-free DMEM for 24 h.
Subsequently, the cells were pretreated with or without NAC (2 mM),
SB203580 (5 μM) or pifithrin-α (5 μM) for 1 h. Next the cells were
treated with different concentrations of IS (0, 25, 50 or 100 μM)
for 24 h. Following incubation, cells were collected and fixed in
70% ethanol for 24 h at 4°C. The cells were centrifuged at 245 × g
for 5 min and the cell pellet was resuspended in 400 μl
phosphate-buffered saline (PBS) containing RNase A (10 mg/ml, 50
μl) and PI (2 mg/ml, 10 μl). The mixture was incubated in the dark
at 37°C for 30 min and then analyzed using a FACSCalibur™ cytometer
(BD Biosciences, San Jose, CA, USA). The cell cycle and cell death
data were analyzed using FlowJo software V6.0 (Tree star, Ashland,
OR, USA). The relative DNA content per cell was obtained by
measuring the fluorescence of the DNA. The extent of cell death was
determined by evaluating the sub G1 fraction, or the percentage of
cells with DNA content <2N. The data were replicated three
times.
Western blot assays
A375 cells were pretreated with or without NAC (2
mM), SB203580 (5 μM) or pifithrin-α (5 μM) for 1 h. This was
followed by treatment with different concentrations of IS (0, 25,
50 and 100 μM) for 24 h. The cells were resuspended in lysis buffer
(150 mmol/l NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, and
50 mmol/l Tris-Cl pH 8.0, 2 μg/ml aprotinin, 2 μg/ml leupeptin, 40
mg/ml of phenylmethylsulfonyl fluoride, 2 mmol/l dithiothreitol;
Beyotime Institute of Biotechnology) and centrifuged at 10,080 × g
for 15 min to remove nuclei and cell debris. Supernatants were
frozen at −80°C until use. The protein concentrations were
determined by the Bradford assay (Bio-Rad, Hercules, CA, USA) and
30 μg cellular proteins were electroblotted onto a polyvinylidene
fluoride membrane (Millipore, Billerica, MA, USA) following
separation using 10% SDS-polyacrylamide gel electrophoresis. The
immunoblot was blocked for 1 h with 5% milk at room temperature
followed by an overnight incubation at 4°C with a 1:1,000 dilution
of primary antibodies against P-p38, P-p53, p21, P-CDK1, CDK2,
cyclin E, cyclin B1, cleaved PARP or β-actin. Blots were washed
twice with Tween 20/Tris-buffered saline (TTBS) prior to addition
of a 1:1,000 dilution of horseradish peroxidase-conjugated
secondary antibody for 1 h at room temperature. Blots were again
washed with TTBS [Sangon Biotech (Shanghia) Co., Ltd., Shanghai,
China] before development by enhanced chemiluminescence using
Supersignal West Femto Chemiluminescent substrate (Pierce,
Rockford, IL, USA). Band intensities were quantified using
UN-SCAN-IT Gel Analysis software (version 6; Silk Scientific, Orem,
UT, USA). The optical density for the target protein was shown as a
proportion of the β-actin optical density. The western blot data
were replicated three times.
Evaluation of ROS
ROS were detected using the cell-permeable
fluorescent probe 2,7-dichlorodihydrofluorescein diacetate
(H2DCFDA; Sigma-Aldrich), a non-fluorescent compound,
which is converted into highly fluorescent
dichlorodihydrofluorescein by cellular peroxides. The A375 cells
were exposed to various concentrations of IS (0, 25, 50 and 100 μM)
for 6 h and were then loaded with H2DCFDA (10 μM) in
serum-free DMEM. Following incubation at 37°C for 30 min, cells
were washed with PBS and fluorescence was monitored by flow
cytometry at excitation wavelength of 488 nm and an emission
wavelength of 530 nm. The mean fluorescence intensity (MFI) data
was analyzed using FlowJo software V6.0. The MFI data were
replicated three times.
Statistics
All data are presented as the mean ± standard
deviation. Data analysis was performed by one-way analysis of
variance. For comparison of two groups, a Student’s t-test was
used. P<0.05 was considered to indicate a statistically
significant difference.
Results
IS inhibits cell viability and
proliferation in A375 cells
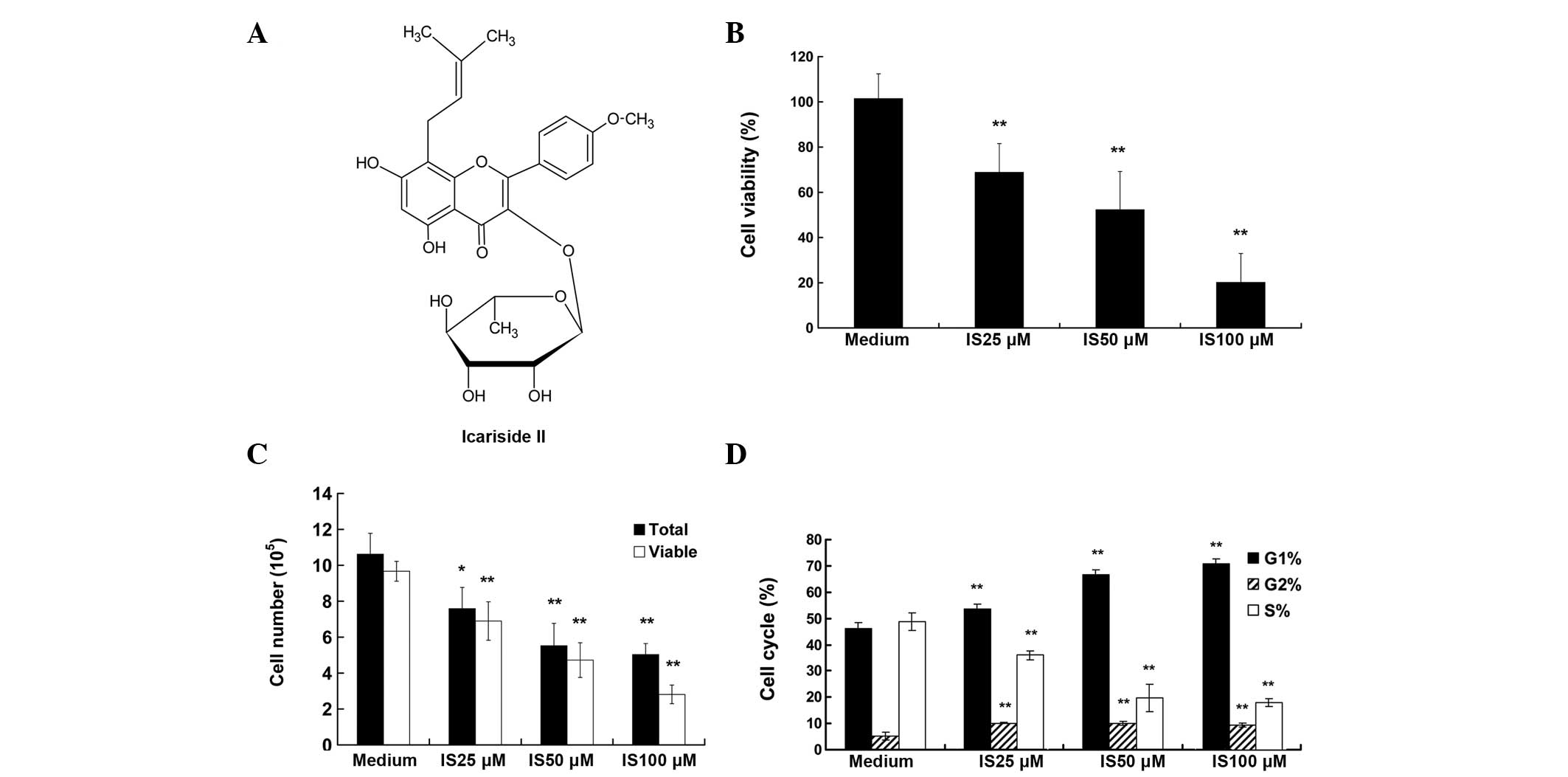
The viability of A375 cells was tested following
treatment with increasing concentrations of IS (0, 25, 50 and 100
μM) for 24 h. As demonstrated by the WST-8 assay, treatment with IS
resulted in markedly reduced cell viability, from 77 to 21% (25 and
100 μM respectively; P<0.01) (Fig.
1B). The detection of living and total cell numbers was
determined by the Casy Cell Counter and Analyzer system. As
displayed in Fig. 1C, following a
24-h incubation period, the total cell numbers in the medium
control group was increased from 2.00×105 to
10.62×105, and IS treatment significantly decreased
total cell numbers, as compared with that of the medium control
group (P<0.01). For example, only 5×105 total cells
were counted in the IS 100 μM treatment group. A similar trend was
observed in the living cells data (P<0.01).
IS induces cell cycle arrest and inhibits
the expression of cell cycle-related proteins in A375 cells
As a reduction in cell proliferation may result from
the induction of cell cycle arrest, the present study investigated
whether the IS-induced growth inhibition was due to cell cycle
arrest. Cell cycle distribution analysis (Fig. 1D) showed that the percentage of
cells in G0/G1 phase increased with the increasing IS concentration
and peaked at 100 μM of IS (69.51%), as compared with that of the
medium control group (44.01%). By contrast, the percentage of cells
in the S phase was reduced accordingly (P<0.01). Additionally,
IS treatment induced G2/M arrest (P<0.01), although to a lesser
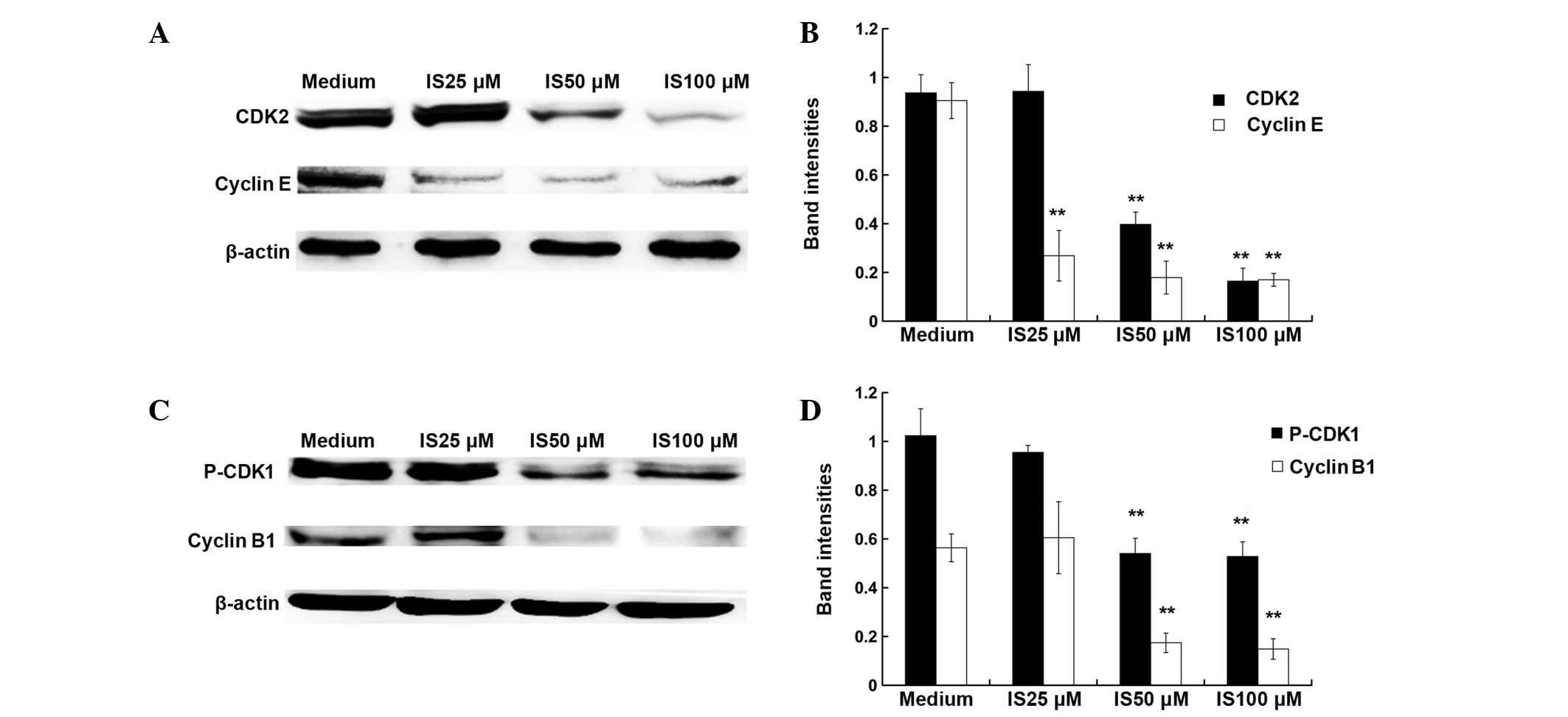
extent. The cell cycle is regulated by cyclins and cyclin-dependent
kinases (20). As demonstrated by
western blot assay (Fig. 2), 50
and 100 μM IS treatment significantly inhibited the expression
levels of cyclin E, CDK2, cyclin B1 and P-CDK1 (P<0.01), while
25 μM IS treatment only caused a significant reduction in the
expression levels of cyclin E (P<0.01).
IS induces the production of ROS and
activates p38, p53 and p21
To investigate the molecular mechanism for
IS-induced cell cycle arrest, the present study examined whether IS
induces the generation of ROS. The levels of ROS were determined 6
h after IS treatment. As shown in Fig.
3A and B, flow cytometry revealed that IS-induced ROS
generation was ~2.3-fold higher compared with that in the medium
controls (25 μM; P<0.01). A previous study has shown that ROS
induce cell cycle arrest via activation of p38, p53 and p21
(20). In the present study,
western blot analysis demonstrated that IS (25, 50 and 100 μM)
treatment significantly increased the phosphorylation of p38 and
p53 compared with that in the medium controls (P<0.01; Fig. 3C and D). An increased expression
level of p21 was also observed following IS treatment compared with
that in the medium controls (P<0.01).
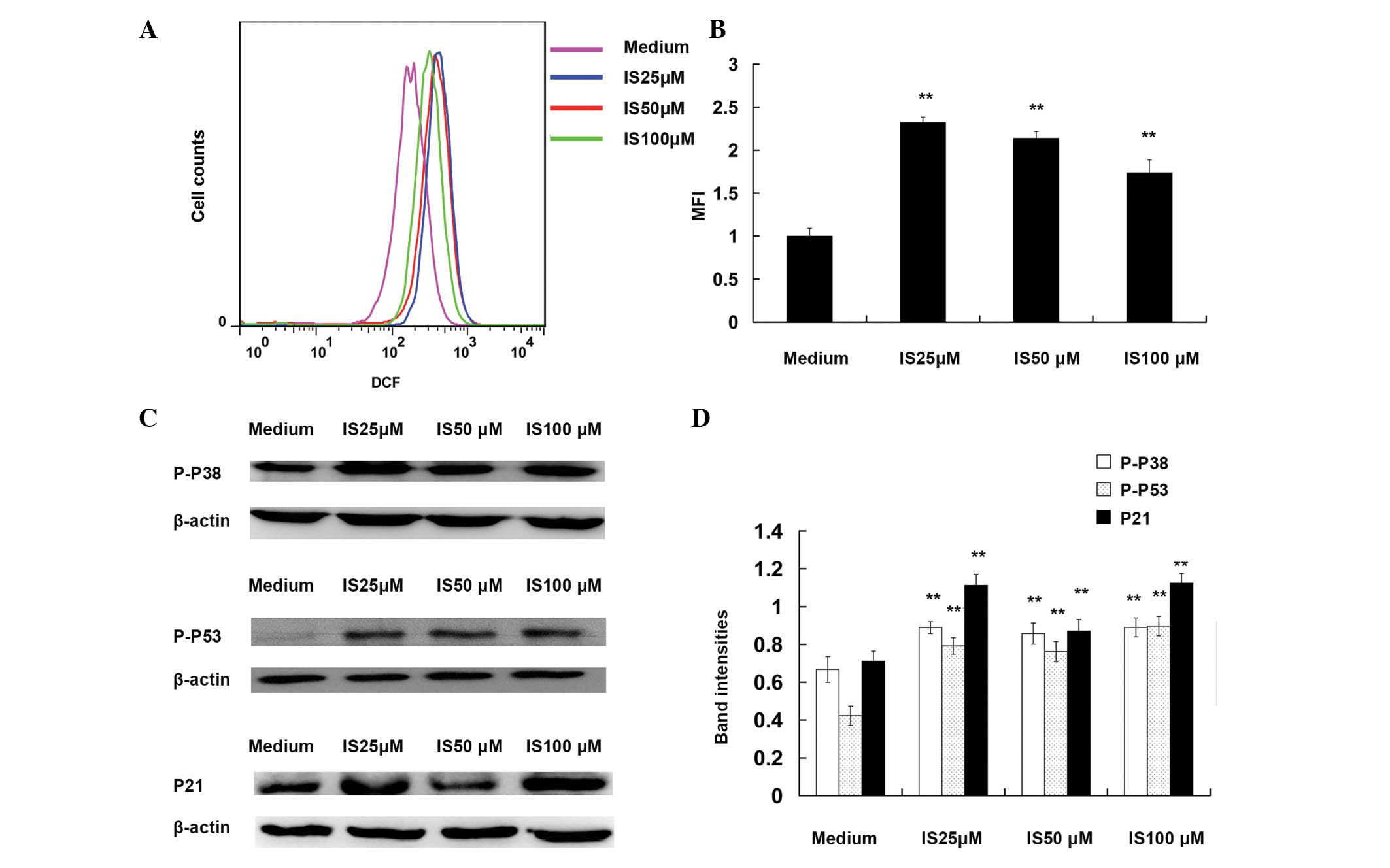 | Figure 3Icariside II (IS) induces the
production of reactive oxygen species (ROS) and activates p38, p53
and p21. For the ROS assay, A375 cells were exposed to various
concentrations of IS (0, 25, 50 and 100 μM) for 6 h and then were
loaded with 2,7-dichlorodihydrofluorescein diacetate (10 μM).
Following incubation at 37°C for 30 min, cells were washed with
phosphate-buffered saline and their fluorescence measured using
either flow cytometry or fluorescence microscopy. The mean
fluorescence intensity (MFI) data were analyzed with FlowJo
software V6.0. (A) Representative images of ROS MFI. (B)
Statistical data of ROS MFI. For western blotting assays, A375
cells were treated with different concentrations of IS (0, 25, 50
and 100 μM) for 24 h. The total protein were extracted, and the
levels of P-p38, P-p53 and p21 were detected by western blot
analysis. β-actin was used as the loading control. (C)
Representative western blot images of P-p38, P-p53 and p21. (D)
Quantification of band intensities of P-p38, P-p53, and p21. Band
intensities were quantified using UN-SCAN-IT Gel Analysis software.
**P<0.01, as compared with medium control group. |
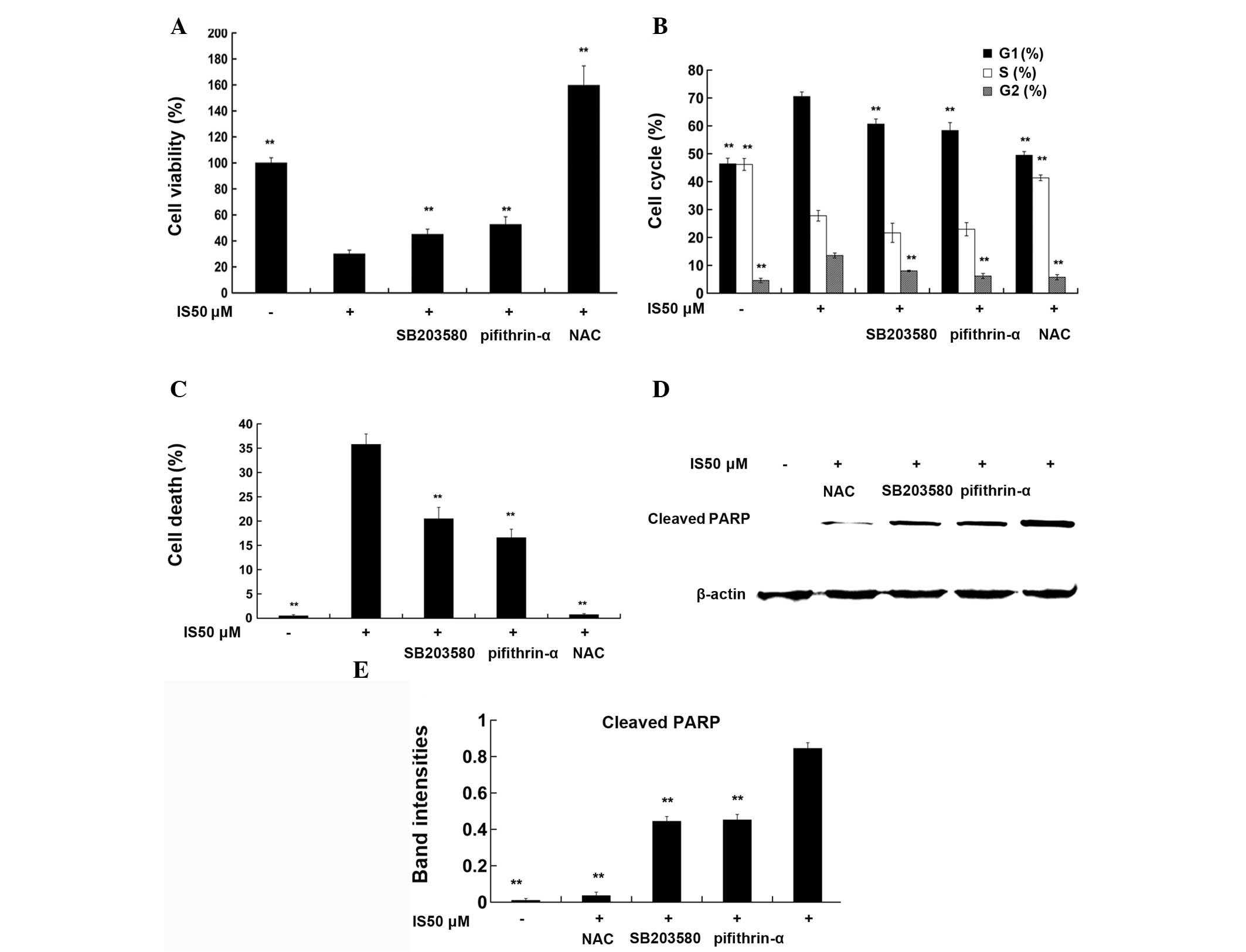
NAC, SB203580, and pifithrin-α reverse
the effects of IS on cell viability, cell cycle arrest and cell
death
As demonstrated by WST-8 assay (Fig. 4A), treatment with 50 μM IS for 24 h
significantly reduced cell viability compared with that in the
medium controls (P<0.01). Pretreatment with NAC (2 mM), a ROS
scavenger, for 1 h completely reversed the IS-mediated reduction in
cell viability (P<0.01), while pretreatment with SB203580 (5
μM), a p38 inhibitor, or pifithrin-α (5 μM), a p53 inhibitor, for 1
h partially reversed the IS-mediated reduction of cell viability
(P<0.01). Cell cycle data (Fig.
4B) demonstrated that 50 μM IS treatment induced G0/G1 phase
and G2/M phase arrest compared with that in the medium controls
(P<0.01), while pretreatment with NAC, SB203580 or pifithrin-α
reversed IS-induced cell cycle arrest, respectively (P<0.01).
Fig. 4C shows that treatment with
IS 50 μM for 24 h resulted in a marked increase in the levels of
cell death (33.60%) compared with those in the medium control
(0.26%). NAC, SB203580 or pifithrin-α pretreatment partly reversed
the IS-mediated increase in the levels of cell death (P<0.01).
Similar trends were observed in the levels of cleaved PARP, a
marker of cells undergoing apoptosis (Fig. 4D and E).
Discussion
Since deregulated proliferation and inhibition of
apoptosis are key processes in the development of all types of
tumor, they present two clear targets for therapeutic intervention
in tumors (21). In the current
study, the cell counting data demonstrated that IS markedly
inhibited cell proliferation in A375 melanoma cells. The rate of
cell proliferation is also identified through the calculation of
the proportion of cells in various phases of the cell cycle, with
the easiest being the S phase (22). The cell cycle data showed that IS
significantly reduced the proportion of A375 melanoma cells in the
S phase, which further supports the anti-proliferative effects of
IS.
Uncontrolled cellular proliferation is the result of
cell cycle disorganization (22).
Numerous families of regulatory proteins possess key roles in the
control of cell cycle progression, including the cyclins, CDKs,
their substrate proteins, the CDK inhibitors and the
tumor-suppressor gene products p53 and pRb (20). These families comprise the basic
regulatory machinery responsible for catalyzing cell cycle
transition. For example, the cyclin E-CDK2 complex has a critical
role in the G1/S phase transition (23) and the cyclin B1-CDK1 complex is
expressed predominantly during G2/M phase (24). Data from the present study showed
that IS-induced cell cycle arrest of A375 cells predominantly
occurs during the G0/G1 phase, occurring to a lesser extent during
the G2/M phase. These effects were mediated by inhibition of cell
cycle-related proteins, such as cyclin E, CDK2, cyclin B1 and CDK1.
A previous study reported that IS could potentiate
paclitaxel-induced cell death in A375 human melanoma cells
(25). Paclitaxel mainly induces
the cell cycle blockage at the G2/M boundary (26,27),
while IS mainly induces the cell cycle blockage at the G1/S
boundary. The synergistic mechanism may be attributed to different
cell cycle regulation of these two compounds.
Evidence from a previous study implies that there is
a positive correlation between cellular redox status and cytotoxic
efficacy of anti-cancer agents (28). The present study reported that IS
inhibited the cell viability and cell proliferation through the
generation of ROS. These findings were further supported by the
evidence that pretreatment with NAC blocked IS-mediated reduction
of cell viability, increase of cell death and cell cycle arrest.
Following IS treatment (6 h), the ROS levels in the 100 μM IS
treatment group were lower than those in the 50 and 25 μM IS
treatment groups. These ROS data were inconsistent with the cell
viability and proliferation data. Perhaps the time elapsed for ROS
production to reach a peak level following treatment with 100 μM
IS, is significantly less than that after treatment with 25 and 50
μM.
A previous study has indicated that ROS induces the
activation of the MAPK pathways and that the activation of p38 MAPK
is involved in cell growth arrest (24). p38 MAPK activation leads to
accumulation of p53 by directly phosphorylating it at selected
amino acid residues (29). p53
protein is an important cell cycle check-point regulator at the
G1/S and G2/M check points (30,31).
p53-dependent cell cycle arrest is mainly mediated by
transcriptional activation of p21 (24). In this study, it was revealed that
IS treatment induced massive ROS accumulation in A375 cells, and
activated a series of important related proteins, including p38,
p53 and p21. These results were further supported by the evidence
that pretreatment with SB203580 or pifithrin-α significantly
blocked IS-mediated reduction of cell viability, increase of cell
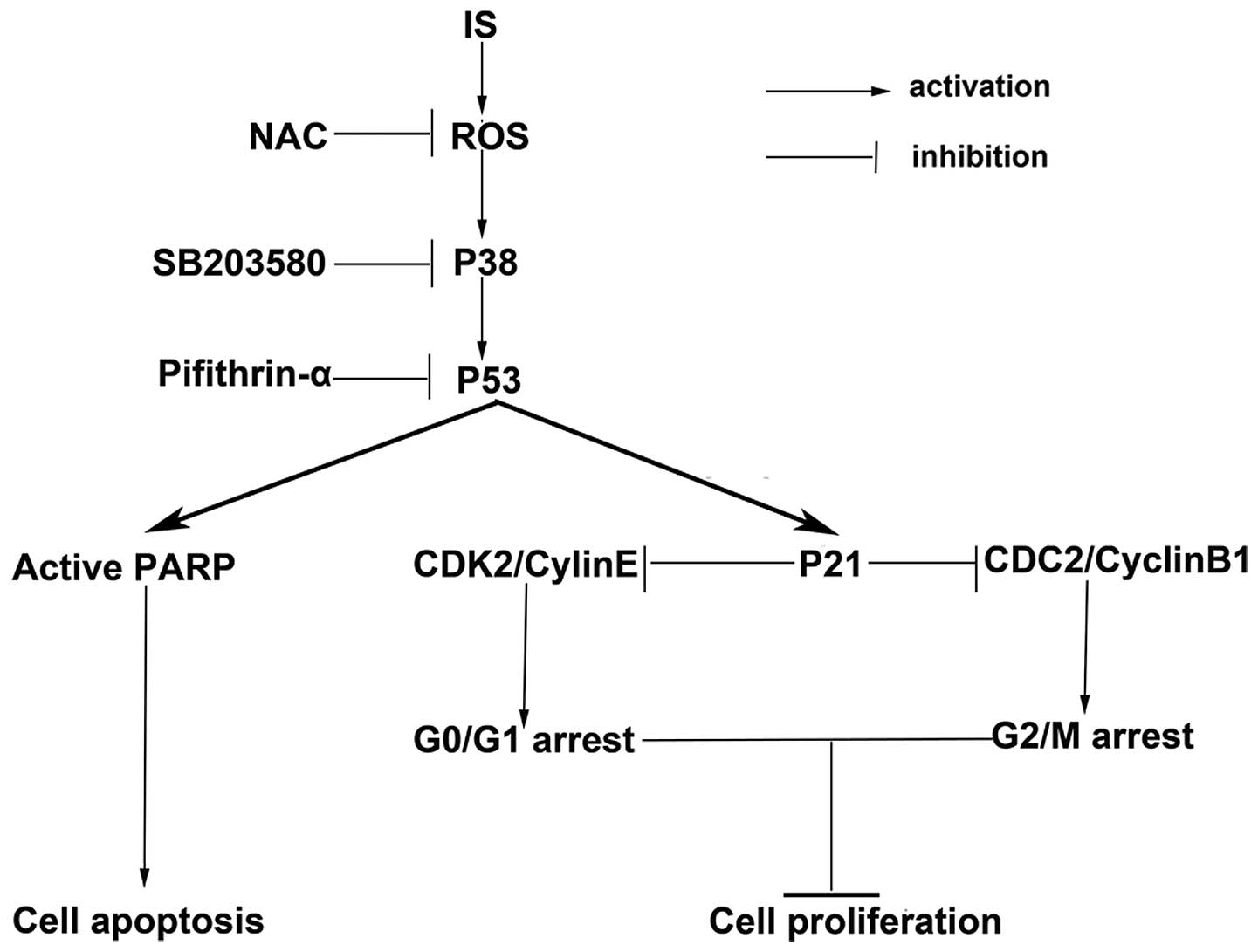
death and cell cycle arrest. Therefore, it was determined that the
ROS-mediated p38-p53 signaling pathway was involved in IS-induced
reduction of cell viability and cell proliferation, increase of
cell death and cell cycle arrest (Fig.
5).
In the present study, SB203580 and pifithrin-α
pretreatment failed to completely reverse IS-induced cell death and
cell cycle arrest. These findings suggested that other important
mechanisms may be involved in IS-mediated cytotoxic effects. A
previous study demonstrated that IS sensitized U937 acute myeloid
leukemia cells to apoptosis via activating JAK2-STAT3 signaling
(20). Further studies are
necessary to investigate the association between IS-mediated
activation of the ROS-p38-p53 signaling pathway and inactivation of
the JAK2-STAT3 signaling pathway in human melanoma cells.
In conclusion, the present study reports that IS
inhibits cell proliferation and induces cell cycle arrest.
Additionally, it confirms that these effects are mediated at least
in part by activation of the ROS-p38-p53 signaling pathway. These
findings suggest that IS may be a potential chemotherapeutic agent
in treatment of melanoma in the future.
Acknowledgements
This study was funded by a grant from the National
Natural Science Foundation of China (81102541).
References
|
1
|
Rigel DS, Russak J and Friedman R: The
evolution of melanoma diagnosis: 25 years beyond the ABCDs. CA
Cancer J Clin. 60:301–316. 2010.PubMed/NCBI
|
|
2
|
Balch CM, Gershenwald JE, Soong SJ, et al:
Final version of 2009 AJCC melanoma staging and classification. J
Clin Oncol. 27:6199–6206. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Soengas MS and Lowe SW: Apoptosis and
melanoma chemoresistance. Oncogene. 22:3138–3151. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Serrone L, Zeuli M, Sega FM and Cognetti
F: Dacarbazine-based chemotherapy for metastatic melanoma:
thirty-year experience overview. J Exp Clin Cancer Res. 19:21–34.
2000.PubMed/NCBI
|
|
5
|
Tsao H, Atkins MB and Sober AJ: Management
of cutaneous melanoma. N Engl J Med. 351:998–1012. 2004. View Article : Google Scholar
|
|
6
|
Panka DJ, Atkins MB and Mier JW: Targeting
the mitogen-activated protein kinase pathway in the treatment of
malignant melanoma. Clin Cancer Res. 12:2371s–2375s. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Boutros T, Chevet E and Metrakos P:
Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase
regulation: roles in cell growth, death, and cancer. Pharmacol Rev.
60:261–310. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dolado I, Swat A, Ajenjo N, De Vita G,
Cuadrado A and Nebreda AR: p38alpha MAP kinase as a sensor of
reactive oxygen species in tumorigenesis. Cancer Cell. 11:191–205.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Assefa Z, Vantieghem A, Garmyn M, et al:
p38 mitogen-activated protein kinase regulates a novel,
caspase-independent pathway for the mitochondrial cytochrome c
release in ultraviolet B radiation-induced apoptosis. J Biol Chem.
275:21416–21421. 2000. View Article : Google Scholar
|
|
10
|
Bulavin DV, Demidov ON, Saito S, et al:
Amplification of PPM1D in human tumors abrogates p53
tumor-suppressor activity. Nat Genet. 31:210–215. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vogelstein B, Lane D and Levine AJ:
Surfing the p53 network. Nature. 408:307–310. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xiao D, Powolny AA, Moura MB, et al:
Phenethyl isothiocyanate inhibits oxidative phosphorylation to
trigger reactive oxygen species-mediated death of human prostate
cancer cells. J Biol Chem. 285:26558–26569. 2010. View Article : Google Scholar
|
|
13
|
Xiao D, Powolny AA and Singh SV: Benzyl
isothiocyanate targets mitochondrial respiratory chain to trigger
reactive oxygen species-dependent apoptosis in human breast cancer
cells. J Biol Chem. 283:30151–30163. 2008. View Article : Google Scholar
|
|
14
|
Lee KS, Lee HJ, Ahn KS, et al:
Cyclooxygenase-2/prostaglandin E2 pathway mediates icariside II
induced apoptosis in human PC-3 prostate cancer cells. Cancer Lett.
280:93–100. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim SH, Ahn KS, Jeong SJ, et al: Janus
activated kinase 2/signal transducer and activator of transcription
3 pathway mediates icariside II-induced apoptosis in U266 multiple
myeloma cells. Eur J Pharmacol. 654:10–16. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kang SH, Jeong SJ, Kim SH, et al:
Icariside II induces apoptosis in U937 acute myeloid leukemia
cells: role of inactivation of STAT3-related signaling. PloS one.
7:e287062012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xia Q, Xu D, Huang Z, Liu J, Wang X and
Liu S: Preparation of icariside II from icariin by enzymatic
hydrolysis method. Fitoterapia. 81:437–442. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Röhner E, Kolar P, Seeger JB, et al:
Toxicity of antiseptics against chondrocytes: what is best for the
cartilage in septic joint surgery? Int Orthop. 35:1719–1723.
2011.PubMed/NCBI
|
|
19
|
Röhner E, Matziolis G, Perka C, et al:
Inflammatory synovial fluid microenvironment drives primary human
chondrocytes to actively take part in inflammatory joint diseases.
Immunol Res. 52:169–175. 2012.PubMed/NCBI
|
|
20
|
Gali-Muhtasib H and Bakkar N: Modulating
cell cycle: current applications and prospects for future drug
development. Curr Cancer Drug Targets. 2:309–336. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Evan GI and Vousden KH: Proliferation,
cell cycle and apoptosis in cancer. Nature. 411:342–348. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Golias CH, Charalabopoulos A and
Charalabopoulos K: Cell proliferation and cell cycle control: a
mini review. Int J Clin Pract. 58:1134–1141. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ma T, Van Tine BA, Wei Y, et al: Cell
cycle-regulated phosphorylation of p220(NPAT) by cyclin E/Cdk2 in
Cajal bodies promotes histone gene transcription. Genes Dev.
14:2298–2313. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kawamoto H, Koizumi H and Uchikoshi T:
Expression of the G2-M checkpoint regulators cyclin B1 and cdc2 in
nonmalignant and malignant human breast lesions: immunocytochemical
and quantitative image analyses. Am J Pathol. 150:15–23. 1997.
|
|
25
|
Wu J, Guan M, Wong PF, Yu H, Dong J and Xu
J: Icariside II potentiates paclitaxel-induced apoptosis in human
melanoma A375 cells by inhibiting TLR4 signaling pathway. Food Chem
Toxicol. 50:3019–3024. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Schiff PB and Horwitz SB: Taxol stabilizes
microtubules in mouse fibroblast cells. Proc Natl Acad Sci USA.
77:1561–1565. 1980. View Article : Google Scholar
|
|
27
|
Crossin KL and Carney DH: Microtubule
stabilization by taxol inhibits initiation of DNA synthesis by
thrombin and by epidermal growth factor. Cell. 27:341–350. 1981.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Engel RH and Evens AM: Oxidative stress
and apoptosis: a new treatment paradigm in cancer. Front Biosci.
11:300–312. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bulavin DV, Saito S, Hollander MC, et al:
Phosphorylation of human p53 by p38 kinase coordinates N-terminal
phosphorylation and apoptosis in response to UV radiation. EMBO J.
18:6845–6854. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
She QB, Chen N and Dong Z: ERKs and p38
kinase phosphorylate p53 protein at serine 15 in response to UV
radiation. J Biol Chem. 275:20444–20449. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kastan MB, Canman CE and Leonard CJ: P53,
cell cycle control and apoptosis: implications for cancer. Cancer
Metastasis Rev. 14:3–15. 1995. View Article : Google Scholar
|