Introduction
Ischemic heart disease is the primary cause of
fatalities in developed countries, contributing to ~40% of total
mortality (1). One type of
ischemic heart disease condition, ischemia/reperfusion (IR) injury,
induces myocardiocyte apoptosis and necrosis, and contributes to
25% of the total mortality in patients with acute myocardial
infarction (2). In addition, IR
injury results in the degradation of excess autophagic flux, and
the recycling of misfolded proteins or dysfunctional organelles
(3). With a ‘housekeeper’
function, autophagy is critical in maintaining cellular homeostasis
and mediating resistance to apoptosis or senescence (4). Although the protective role of
autophagy is widely known, the adaptive autophagic mechanism during
myocardiocyte survival remains controversial (5). For example, the induction of
autophagy through autophagy-related gene 7 (Atg7) overexpression
was shown to be protective in the heart during ischemia (6). However, activated autophagy has been
associated with aggravated cardiac hypertrophy in pressure
overload-induced heart failure (7). Recently, adenosine and acetylcholine
have been shown to provide cardioprotective effects through an
underlying autophagy-dependent mechanism (8,9).
Thus, autophagy may be required for myocardiocyte survival during
ischemia.
Lycopene (Ly), the most common type of carotenoid in
dietary intake, has been shown to confer protective effects against
ageing, tissue damage, certain types of cancer, atherosclerosis and
associated coronary artery disease (10). Possible mechanisms of Ly-mediated
effects are via antioxidant activity, increases in gap-junctional
intercellular communication, cell growth control, the activation of
the mevalonic acid pathway, suppressing the expression of the
oncogene ras and the modulation of immune responses
(11–14). However, the impact of Ly on IR
injury induced by myocardiocyte apoptosis and necrosis has been
poorly investigated. During IR, the balance between reactive oxygen
species (ROS) generation and elimination is critical in determining
cell survival (15). Furthermore,
the deregulation of ROS directly induces autophagy in various
stress conditions (16).
Therefore, Ly may induce autophagy to switch from the cell death
pathway to survival in hypoxia/reoxygenation (HR)-induced H9C2
myocardioblast cells.
The present study was conducted to investigate
whether the autophagy induced by Ly is associated with HR-induced
H9C2 myocardioblast cell damage and whether the proautophagic
effect of Ly is involved in the cell protection mechanism.
Materials and methods
Reagents
Dulbecco’s modified Eagle’s medium (DMEM), fetal
bovine serum (FBS) and Lipofectamine RNAiMax were purchased from
Invitrogen (Carlsbad, CA, USA). Microtubule-associated protein
1-light chain 3β (MAP1LC3B), BECN1 and negative
control (NC) small interfering (si)RNA were synthesized by
GenePharma (Shanghai, China). The chemical reagents compound C
(CC), Ly, chloroquine and
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide
(MTT), were obtained from Sigma-Aldrich (St. Louis, MO, USA). The
cell apoptosis detection kit was purchased from BD Biosciences
(Franklin Lakes, NJ, USA). Rabbit monoclonal antibodies specific to
LC3 (2057-1; 1:5000), phospho-adenosine monophosphate kinase
(p-AMPK, 2802-1, 1:1000), AMPK (1596-1, 1:10000), Bax (1063-1,
1:5000), B-cell lymphoma 2 (Bcl-2, 1017-1, 1:1000 ) and β-actin
(5779-1, 1:10000) and the horseradish peroxidase (HRP) conjugated
goat anti-rabbit immunoglobulin (Ig) G secondary antibody (3053-1,
1:10000) were obtained from Epitomics (Burlingame, CA, USA). Rabbit
monoclonal antibodies specific to Beclin 1 (3738, 1:1000) and
active-caspase-3 (9660, 1:1000) were purchased from Cell Signaling
Technology (Denver, MA, USA).. The H9C2 cell line was donated by
the Pharmacological Laboratory of Zhejiang University (Hangzhou,
China).
Cell culture
The H9C2 cells were grown to 80% confluence in DMEM
containing 10% FBS at 37°C in a humidified 5% CO2/95%
air atmosphere. Subsequent to starvation in serum-free DMEM for 6 h
in a normal atmosphere, the H9C2 cells were immediately incubated
in serum- and glucose-free DMEM and were moved into a hypoxic
atmosphere containing 94% N2, 5% CO2 and 1% O2 to mimic ischemia..
After 16 h of hypoxia, the cells were rapidly transferred into a
normal atmosphere with DMEM containing 10% FBS and glucose for
reoxygenation (2 h). Prior to the HR, the H9C2 cells were treated
with MAP1LC3B, BECN1 or NC siRNA, or Ly, with
or without chloroquine, for 48 h, and 1 μM CC was added for 24 h
prior to HR to suppress AMPK phosphorylation.
siRNA transfection
The following siRNA sequences were transfected into
the cells: MAP1LC3B sense, 5′-CUCCCUAAGAGGAUCUUUATT-3′;
MAP1LC3B antisense, 5′-UAAAGAUCCUCUUAGGGAGTT-3′;
BECN1 sense, 5′-GUGGAAUGGAAUGAGAUUATT-3′; BECN1
antisense, 5′-UAAUCUCAUUCCAUUCCACTT-3′; NC sense,
5′-UUCUCCGAACGUGUCACGUTT-3′; and NC antisense,
5′-ACGUGACACGUUCGGAGAATT-3′. The H9C2 cells were transfected with
the respective siRNAs according to the instructions provided by the
manufacturers of Lipofectamine RNAiMax.
Cell viability assay
The H9C2 cells were seeded in 96-well plates.
Following HR, 5 mg/ml MTT solution was added and the cells were
incubated for 4 h at 37°C. The supernatants were then removed and
the cells were solubilized with 150 μl dimethyl sulfoxide (Shanghai
Shengong Biotechnology Co., Shanghai, China). The absorbance at 490
nm was measured using an ELISA plate reader (Model 550; Bio-Rad,
Hercules, CA, USA).
Detection of cell apoptosis
Following HR treatment, the cells were harvested and
incubated with 5 μl Annexin V-fluorescein isothiocyanate and 2.5
μg/ml propidium iodide (PI) for 15 min, and then subjected to flow
cytometric analysis (BD FACS; BD Biosciences, Franklin, NJ,
USA).
Western blotting
Subsequent to HR, followed by careful washing, the
H9C2 cells were harvested and lysed on ice for 45 min using
moderate lysis buffer (Beyotime, Haimen, China). Equal quantities
of cell proteins were separated via sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and were then
transferred to polyvinylidene fluoride membranes (Millipore,
Billerica, MA, USA). The membranes were blocked with 5% non-fat dry
milk for 1 h at room temperature and then probed with primary
antibodies overnight at 4°C, then washed with 1× TBST (Shanghai
Shengong Biotechnology Co., Shanghai, China) for 3×10 min.
Following expose to the horse radish peroxidase-conjugated
secondary antibodies for 1 h at room temperature and then washed
with 1× TBST for 3×10 min, immunoreactivity was detected using an
enhanced chemiluminescence reagent (Bio-Max, Haemek, Israel).
Statistical analysis
The data are presented as the means ± standard error
of the mean from a minimum of three experiments and analyzed with a
one-way analysis of variance using SPSS 14.0 for Windows (SPSS,
Inc., Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
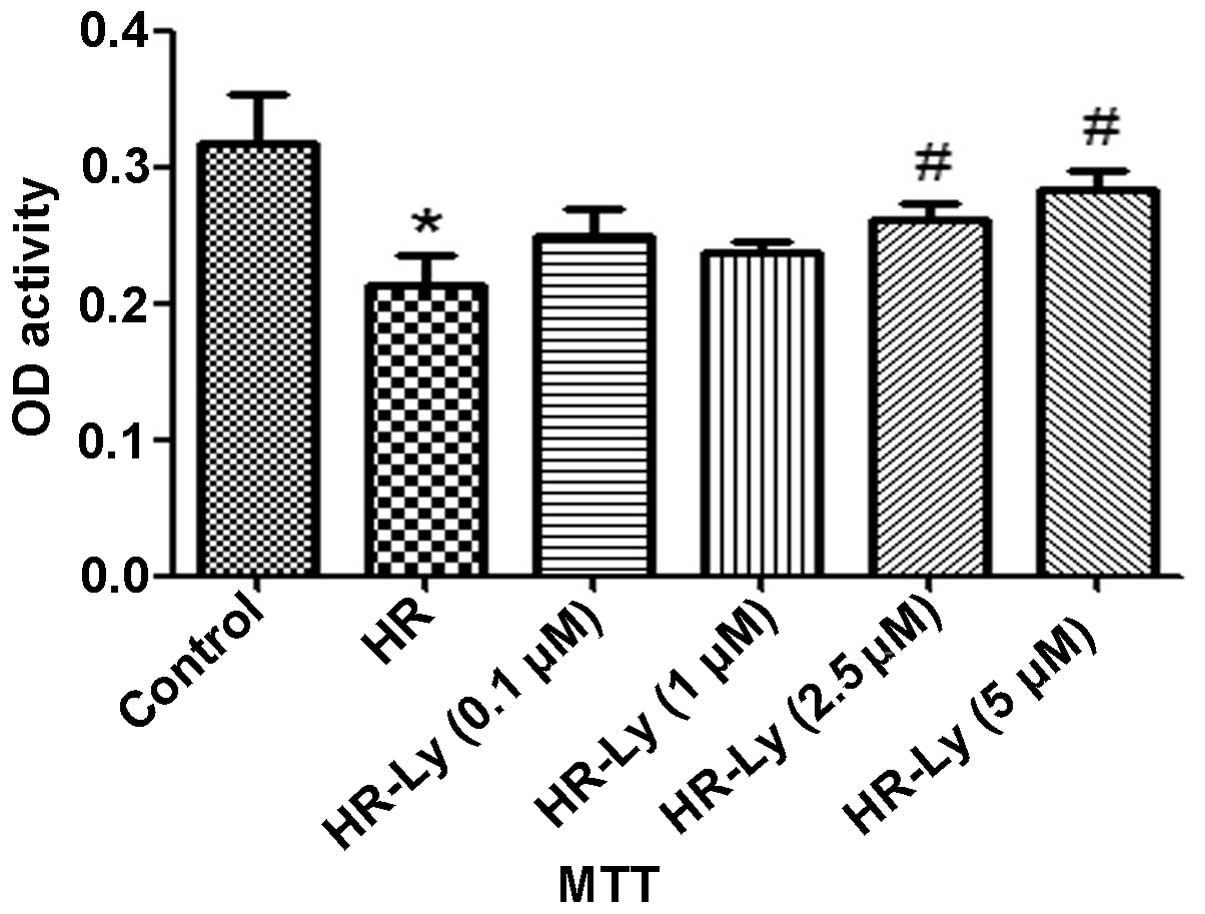
Ly improves HR-induced H9C2 cell
viability
The MTT assay revealed that H9C2 cell viability was
significantly reduced following 16/2 h H/R. Pre-incubation of the
H9C2 cells with Ly resulted in the development of tolerance to HR
in a dose-dependent manner (Fig.
1). These data indicate that Ly protected the H9C2 cells
against HR-induced damage.
Ly reduces HR-induced H9C2 cell
apoptosis
HR in H9C2 cells has been shown to result in an
increase in the rate of apoptosis (9); therefore, whether Ly reduces the rate
of H9C2 cell apoptosis following HR administration was investigated
in the present study. As expected, HR treatment for 16/2 h
significantly increased the percentage of apoptotic cells (Annexin
V-positive, PI-positive and -negative cells) to 35.05%, as compared
with that of the untreated cells (8.48%; P<0.05). Preincubation
of HR-treated cells in Ly-enriched cell-medium significantly
reduced the percentage of apoptotic cells to 21.22 (Ly, 1 μM), 13.9
(Ly, 2.5 μM) and 13.51% (Ly, 5 μM; all P<0.05; Fig. 2A and B), as compared with
HR-treated cells maintained in control medium. To further examine
the association between the inhibition of apoptosis and Ly
treatment, the expression levels of the Bax/Bcl-2 and
active-caspase-3 proteins, commonly used as apoptotic biomarkers,
were detected. The data reveal significantly elevated levels of
Bax/Bcl-2 (3.85-fold) and active-caspase-3 (2.12-fold) in the
HR-treated cells (P<0.05); however, the Bax/Bcl-2 and
active-caspase-3 protein expression levels in the HR-treated cells
were reduced following the addition of Ly (Fig. 2C and D). These data indicate that
Ly protected the H9C2 cells against HR-induced apoptosis.
 | Figure 2Effect of HR with and without
lycopene treatment on H9C2 myocardioblast cell apoptosis. (A) Flow
cytometric analysis of representative samples from control cells,
HR cells and Ly-pre-incubated cells. Annexin V-positive,
PI-positive and -negative cells were identified as apoptotic. (B)
Quantitative analysis of the percentage of apoptotic cells during
HR following Ly treatment. Ly efficiently attenuated HR-induced
H9C2 cell apoptosis. (C) Effect of Ly co-treatment on apoptotic
biomarker protein expression levels during HR, as assessed by
immunoblot analysis (active-caspase-3, 19 and 17 kDa). (D)
Quantitative analysis of the apoptotic biomarker protein expression
levels. *P<0.05, as compared with control cells;
#P<0.05, as compared with HR cells. HR,
hypoxia/reoxygenation; Ly, lycopene; PI, propidium iodide; Bcl-2,
B-cell lymphoma 2. |
Ly increases autophagy in HR-induced H9C2
cells
As autophagy mediates resistance to apoptosis in
stress conditions (4), the effect
of Ly on autophagy during HR was investigated. Immunoblotting
images revealed significantly lower levels of LC3II/LC3I
(0.58-fold) and Beclin 1 (0.44-fold) in HR cells, as compared with
control cells (P<0.05); however, the levels of these biomarkers
in HR-induced cells were elevated by the addition of Ly in a
concentration-dependent manner (Fig.
3A and B). An increase in LC3II/LC3I has been previously shown
to reflect the impaired fusion of autophagosomes with lysosomes
(4). In the present study, to
accurately evaluate autophagic flux, chloroquine was used to
inhibit autophagolysosomal degradation. Administration of
chloroquine prior to HR treatment caused a further reduction in the
LC3II/LC3I ratio, a 0.39-fold reduction from the control cell
value, as compared with 0.58-fold reduction in HR-induced cells
without chloroquine treatment (Fig. 3A
and B). However, the addition of Ly with chloroquine increased
the LC3II/LC3I ratio in HR-induced cells. These data indicate that
Ly induces autophagic flux upregulation in HR-induced H9C2
cells.
MAP1LC3B or BECN1 knockdown enhances
apoptosis in HR-induced H9C2 cells
Autophagy biogenesis is initiated by the activation
of the Unc-51-like kinase 1 (ULK1) complex and the recruitment of
the Beclin 1 complex, and the “isolation membrane” elongates under
the LC3-phosphatidylethanolamine (PE) complex (17). To investigate the potential
protective effect of autophagy on HR-induced H9C2 cells,
MCP1LC3B or BECN1 siRNA was used to inhibit
autophagic flux. As expected, MCP1LC3B and BECN1
siRNA effectively inhibited LC3 and Beclin 1 protein expression,
respectively (Fig. 4A). Following
incubation with MCP1LC3B or BECN1 siRNA, the H9C2
cells were subjected to HR. When autophagy was activated with the
addition of Ly, the percentage of apoptotic cells was significantly
reduced, as compared with the HR treatment only cells (P<0.05;
Fig. 4B and C). When autophagy was
inhibited with MCP1LC3B or BECN1 siRNA, the
protective effect of Ly against apoptosis disappeared. To further
analyze the cytoprotective effect of autophagy, Bax and
active-caspase-3 protein expression levels were examined. Notably,
autophagic deficiency abolished Ly treatment-mediated inhibition of
Bax/Bcl-2 expression and also significantly increased the levels of
active-caspase-3 during HR, as compared with those in cells treated
with Ly only (Fig. 4D and E).
These data indicate that autophagy protects H9C2 cells against
apoptosis during HR following Ly administration.
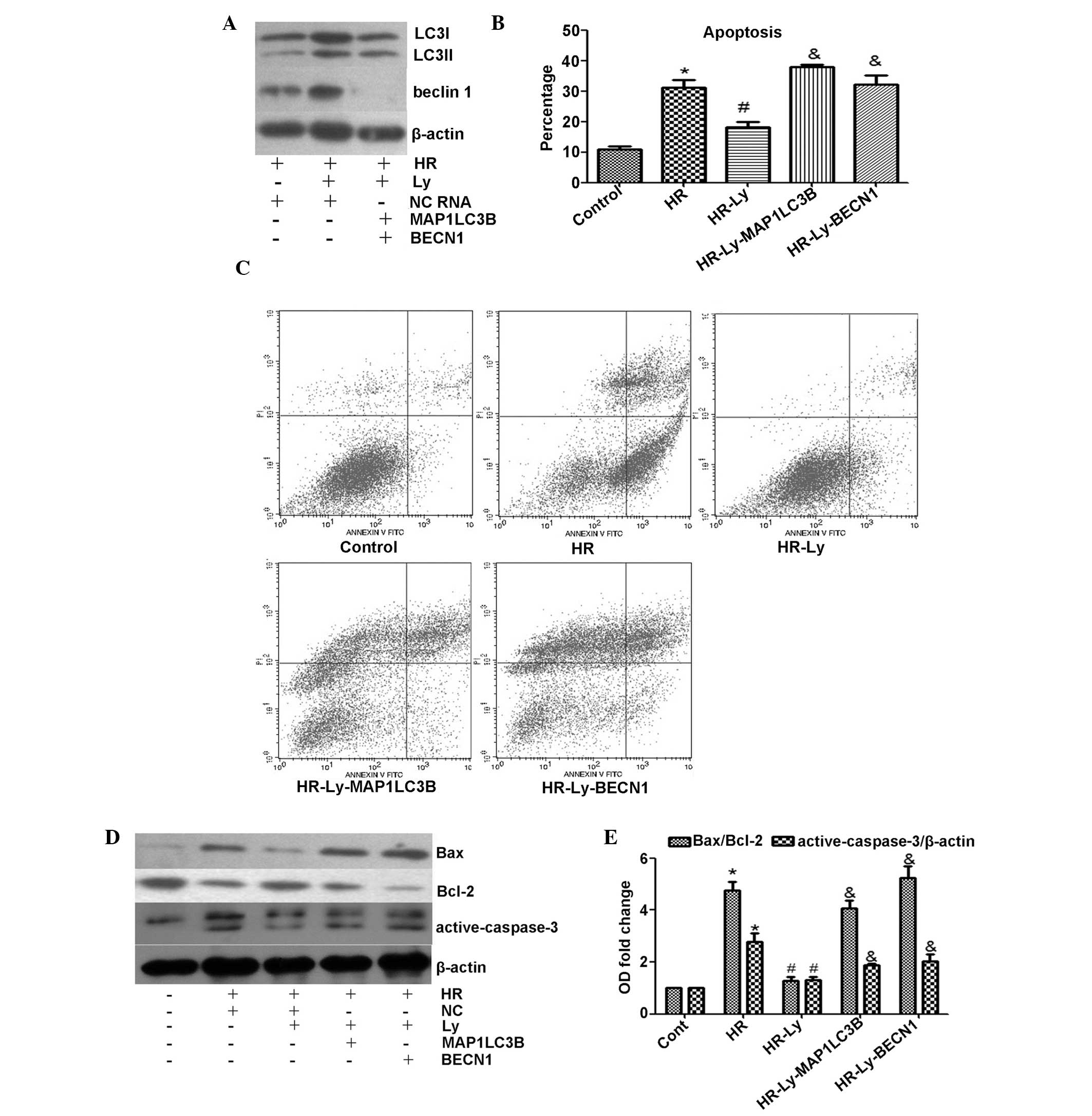 | Figure 4Effect of MCP1LC3B or
BECN1 siRNA on lycopene-induced cytoprotection. The
administration of 2.5 μM Ly was previously demonstrated to provide
maximum cytoprotection; therefore, 2.5 μM Ly was used during the
subsequent experiments. (A) Knockdown efficiency of MCP1LC3B
or BECN1 siRNA was assessed by western blotting (LC3I, 18
kDa; LC3II, 16 kDa). (B) Quantitative analysis of the rate of
apoptosis. Knockdown of LC3 or Beclin 1 abolished lycopene-mediated
cytoprotection during HR. (C) Representative flow cytometric
analysis images of control cells, HR cells, Ly-pre-incubated cells
and siRNA-co-treated Ly cells. (D) Knockdown of LC3 or Beclin 1
exacerbated HR-induced apoptotic biomarker upregulation, as
assessed by western blotting (active-caspase-3, 19 and 17 kDa). (E)
Quantitative analysis of the apoptotic biomarker protein expression
levels. *P<0.05 compared with control cells;
#P<0.05 compared with HR cells;
&P<0.05 compared with HR-Ly cells. MAP1LC3B,
microtubule-associated protein 1-light chain 3β; BECN1, Beclin 1;
siRNA, small interfering RNA; Ly, lycopene; HR,
hypoxia/reoxygenation; LC3, light chain 3; NC, negative control
RNA. |
Suppression of AMPK phosphorylation
induces autophagic inhibition and cell apoptosis
AMPK is known to activate autophagy in a
ULK1-dependent manner. AMPK phosphorylation initiates site specific
phosphorylation of ULK1 and subsequently induces formation of the
ULK1-complex (17). The data from
the present study indicated significantly reduced AMPK
phosphorylation in HR-treated H9C2 cells, as compared with control
cells (P<0.05), although the levels of AMPK phosphorylation in
the HR cells were higher following Ly administration (Fig. 3). To determine whether AMPK
phosphorylation is responsible for Ly-induced autophagy, AMPK
phosphorylation was suppressed using CC. The CC treatment
significantly reduced AMPK phosphorylation (P<0.05; Fig. 5A). In addition, as compared with
control HR and Ly-treatment, the addition of CC significantly
reduced the LC3II/LC3I ratio (P<0.05), induced significantly
higher Bax/Bcl-2 and activate-caspase-3 expression levels
(P<0.05) and significantly increased the apoptotic cell
percentage (P<0.05) upon HR and Ly administration (Fig. 5A and B). The ULK1-complex and
Beclin 1-complex exhibit different regulation mechanisms, as AMPK
phosphorylation has been demonstrated to not affect Beclin 1
expression (17). In the present
study, the Beclin 1 expression levels were not altered upon CC
stress (Fig. 5A). These results
indicate that AMPK phosphorylation-mediated autophagy exerted
cytoprotective effects in HR-induced H9C2 cells following Ly
treatment.
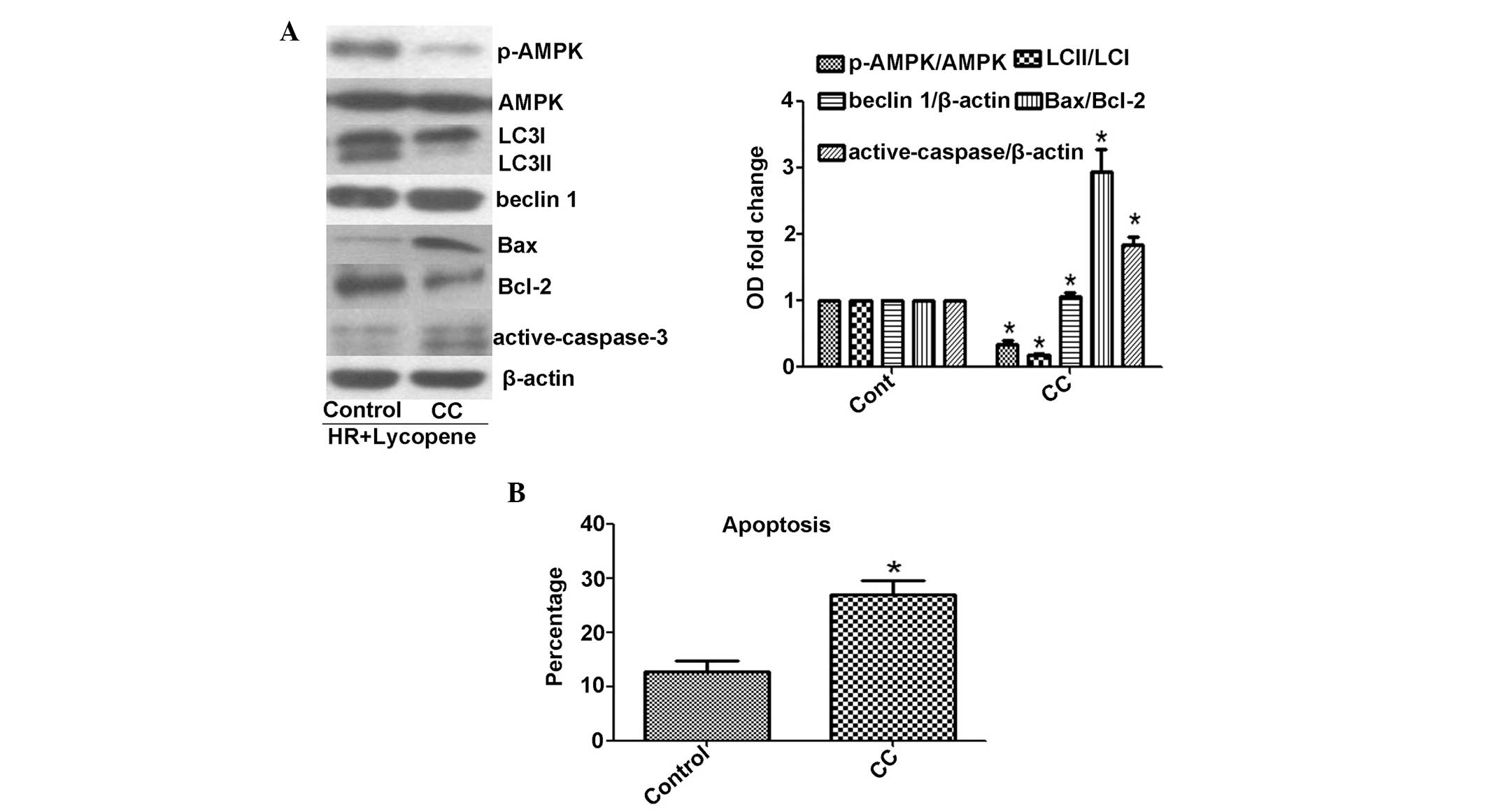 | Figure 5Effect of 1 μM CC on AMPK
phosphoyrlation and lycopene-mediated autophagy and cytoprotection.
Lycopene at 2.5 μM provides cytoprotection. (A) CC suppresses AMPK
phosphorylation, and causes a reduction in LC3 protein expression
levels (LC3I, 18 kDa; LC3II, 16 kDa) and an increase in Bax/Bcl-2
and active-caspase-3 expression levels (active-caspase-3, 19 and 17
kDa). (B) CC induces apoptosis upon HR-lycopene treatment.
*P<0.05, as compared with control cells treated with
HR and lycopene. CC, compound C; AMPK, adenosine monophosphate
kinase; Bcl-2, B-cell lymphoma 2; HR, hypoxia/reoxygenation;
p-AMPK, phospho-AMPK; LC3, light chain 3. |
Discussion
In the present study, HR treatment was demonstrated
to suppress autophagic flux and increase the rate of apoptosis in
H9C2 cells. Nevertheless, preincubation with Ly ameliorated
HR-induced H9C2 cell autophagic inhibition and, in turn, reduced
cell apoptosis. The role of autophagic regulation in HR-induced
H9C2 cell damage following Ly administration was further verified
by MCP1LC3B or BECN1 knockdown. In addition, the
prevention of AMPK phosphorylation using CC reduced Ly-induced
autophagy and increased H9C2 cell apoptosis upon HR stress.
Therefore, autophagy exerted a protective effect on HR-induced H9C2
cells following Ly administration and AMPK phosphorylation may be
involved in the regulation of this Ly-induced autophagy.
In response to stimuli, the ULK1 complex and the
Beclin 1 complex produce phosphatidylinositol 3-phosphate, and a
number of Atg proteins are recruited to initiate autophagy.
Subsequently, Atg4 induces LC3 cleavage to LC3I, which becomes
conjugated to PE to form LC3II, a protein critical for the
elongation and closure of the phagophore. LC3II is then
incorporated into the autophagosomal membranes. When the
autophagosome fuses with lysosome and degenerates, LC3II reverts to
LC3I, is released from the autophagolysosome and is available for
future autophagosome formation (17,18).
ULK1 site-specific phosphorylation is crucial for the assembly of
the ULK1 complex and nucleation of the phagophore. AMPK is required
for ULK1 phosphorylation and induction of autophagy. Knockdown of
AMPK results in ULK1 dephosphorylation and autophagy impairment
(16). As a required quality
control mechanism, autophagy promotes cell homeostasis during
stress conditions; however, inappropriate autophagy accelerates
cell death (19). Therefore,
whether autophagy exerts a protective effect in cardiovascular
diseases remains controversial. Basal autophagy allows cells to
constitutively clear damaged organelles and misfolded proteins.
Upon exposure to stressors, such as IR injury and heart failure,
rapidly upregulated autophagy is an adaptive response to prevent
excessive ROS production, the consumption of ATP, the release of
pro-apoptotic factors, the opening of mitochondrial permeability
transition pores, and the induction of necrosis or apoptosis
(20). Reduced survival and
increased cardiac dysfunction have been observed in mice with
hearts deficient in Parkin, a required factor for mitophagy,
following myocardial infarction (21). In addition, the losses of Atg5
(22), a crucial autophagic
initiation factor, and myeloid cell leukemia 1 (23), an analogue of Bcl-2, in adult heart
have been shown to result in deficient autophagy and rapid heart
failure. In clinical patients, lysosome-associated membrane protein
2 deficiency, a critical fusion factor between autophagosomes and
lysosomes, results in a lethal cardiac hypertrophy (24). By contrast, autophagy-mediated
maladaptive effects on cardiac diseases have been reported. During
pressure overload, angiotensin II-mediated autophagy induces
cardiac contractile dysfunction, pathologic remodeling and cardiac
atrophy (25), and during the
IR-injury phase, accumulating evidence has indicated that the
induction of autophagy is harmful for cardiomyocyte survival,
particularly under an AMPK-independent manner (26). AMPK-dependent autophagy also has
been observed to be maladaptive during IR injury. Inhibiting
autophagy by the downregulation of ULK1 or AMPK reduces the size of
myocardial infarction during the reperfusion phase (27). Notably, Beclin 1-dependent
autophagy is usually considered to be harmful during the IR phase.
Beclin 1 commonly binds to antiapoptotic proteins, such as Bcl-2
and Bcl-xL, to prevent its activation. Upon stress, Bcl-2
dissociation with Beclin 1 allows Beclin 1 activation and
autophagic initiation. Matsui et al (28) and Ma et al (29) have reported that Beclin 1 reduction
significantly attenuates the size of myocardial infarcts during the
IR phase in vivo. Beclin 1-dependent autophagy also exerts
protective effects on IR injury; Sala-Mercado et al
(30) observed that increased
expression levels of Beclin 1 and LC3 were associated with reduced
infarct size. Furthermore, deficient Beclin 1 expression and the
administration of 3-methyladenine have been shown to enhance
ischemic injury (31). Increased
Akt, Bcl-2, Beclin 1 and LC3B expression levels contribute to the
resistance to IR injury in the immature heart, whereas a
maturation-associated impairment in IR injury tolerance may be
attributed to a reduction in Akt, Bcl-2, Beclin 1 and LC3B
expression levels (32). In
vitro, although Valentim et al (33) observed that Beclin 1 knockdown
increases cardiomyocyte survival (33), the majority of studies reveal that
Beclin 1-dependent autophagy is adaptive. Knockdown of Beclin 1
results in maladaptive autophagy and enhances cardiomyocytes
impairment during IR injury (30,34).
A possible explanation for this paradox is that different
autophagic responses are generated during IR injury in vivo
and in vitro. During IR injury, the autophagic flux elevates
in vivo (35), whereas
autophagic levels are reduced in vitro (9,34).
In the present study, autophagy was demonstrated to be negatively
correlated with the rate of apoptosis in H9C2 cells during HR, with
and without Ly, and a positive correlation was detected between
autophagy and H9C2 cell survival. Knockdown of LC3 or Beclin 1
suppressed AMPK phosphorylation, increased apoptosis and elevated
the expression levels of apoptotic biomarkers, including Bax/Bcl-2
and active-caspase-3, which are the most widely investigated
biomarkers of cardiovascular cell death (14). These data indicate that autophagy
promotes H9C2 cell survival by providing resistance to apoptosis
during HR.
Ly is the most prevalent type of antioxidant in the
majority of diets; however, the association between Ly and heart
disease remains controversial. An epidemiologic study has revealed
that β-carotene intake has a modest but significant inverse
correlation with the risk of coronary heart disease (CHD), although
a significant correlation between Ly intake and CHD was not
identified (11); this
inefficiency may be due to a low intake of Ly. Sesso et al
(36) observed that higher serum
Ly concentrations reduced the risk of CHD by 50%. This association
was further verified by the Kuopio Ischemic Heart Disease Risk
Factor Study, which demonstrated a three-fold risk increase in
acute coronary events and stroke in men with a low serum Ly
concentration as compared with other men (37). Ly has also been reported to
directly protect H9C2 cells and rat heart against
ischemia/hypoxia-induced apoptosis (14,38).
This protection depends on lipid peroxidation reduction, oxidative
DNA damage alleviation, cholesterol synthesis inhibition, immune
responses and intercellular communication modulation (14). Recently, an increased dietary
intake of Ly has been reported to correlate with increased
autophagy, reduced apoptosis, inactivated caspase-3 and improved
cell survival in H2O2-induced H9C2 cells, as
well as in iron-supplemented rats (13,14).
In the present study, Ly treatment was negatively correlated with
cell survival and apoptosis during HR, and this protective effect
may occur through increased autophagy. Disruption of autophagy
using MCP1LC3B or BECN1 siRNA, or AMPK
phosphorylation inhibition, counteracted Ly-induced cell survival
and apoptosis reduction in H9C2 cells, and exacerbated cell injury
during HR. Furthermore, active-caspase-3 and Bax/Bcl-2 protein
expression levels were reduced following Ly treatment; however,
knockdown of LC3 and Beclin 1 abolished active-caspase-3 and
Bax/Bcl-2 inactivation by Ly. These data indicate that Ly provides
a protective effect by upregulating autophagy against apoptosis to
promote cell survival in H9C2 cells following treatment with
HR.
In conclusion, autophagy may exert a protective
effect against HR-induced H9C2 cell apoptosis. In the present
study, Ly upregulated autophagy in H9C2 cells following HR,
promoted cell survival and significantly prevented HR-induced
apoptosis. Therefore, therapeutic Ly or autophagic induction may
offer a novel method for CHD treatment. In addition, a novel
mechanism for Ly activity in ischemic conditions has been
described, although this mechanism requires further
investigation.
Acknowledgements
This study was supported by the China National
Natural Science Foundation (grant nos. 30470715, 30870939 and
81170242).
References
|
1
|
Menotti A, Kromhout D, Blackburn H,
Fidanza F, Buzina R and Nissinen A: Food intake patterns and
25-year mortality from coronary heart disease: cross-cultural
correlations in the Seven Countries Study. The Seven Countries
Study Research Group. Eur J Epidemiol. 15:507–515. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yellon DM and Hausenloy DJ: Myocardial
reperfusion injury. N Engl J Med. 357:1121–1135. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Givvimani S, Munjal C, Tyagi N, Sen U,
Metreveli N and Tyagi SC: Mitochondrial division/mitophagy
inhibitor (Mdivi) ameliorates pressure overload induced heart
failure. PLoS One. 7:e323882012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Levine B and Klionsky DJ: Development by
self-digestion: Molecular mechanisms and biological functions of
autophagy. Dev Cell. 6:463–477. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xu X, Pacheco BD, Leng L, Bucala R and Ren
J: Macrophage migration inhibitory factor plays a permissive role
in the maintenance of cardiac contractile function under starvation
through regulation of autophagy. Cardiovasc Res. 99:412–421. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pattison JS, Osinska H and Robbins J: Atg7
induces basal autophagy and rescues autophagic deficiency in
CryABR120G cardiomyocytes. Circ Res. 109:151–160. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cao DJ, Wang ZV, Battiprolu PK, Jiang N,
et al: Histone deacetylase (HDAC) inhibitors attenuate cardiac
hypertrophy by suppressing autophagy. Proc Natl Acad Sci USA.
108:4123–4128. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yitzhaki S, Huang C, Liu W, Lee Y,
Gustafsson AB, Mentzer RM Jr and Gottlieb RA: Autophagy is required
for preconditioning by the adenosine A1 receptor-selective agonist
CCPA. Basic Res Cardiol. 104:157–167. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhao M, Sun L, Yu XJ, et al: Acetylcholine
mediates AMPK-dependent autophagic cytoprotection in H9c2 cells
during hypoxia/reoxygenation injury. Cell Physiol Biochem.
32:601–613. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sharma N and Goswami UC: Functioning of
lycopene in mammalian system: A review. Proc Zool Soc. 64:1–7.
2011. View Article : Google Scholar
|
|
11
|
Voutilainen S, Nurmi T, Mursu J and
Rissanen TH: Carotenoids and cardiovascular health. Am J Clin Nutr.
83:1265–1271. 2006.PubMed/NCBI
|
|
12
|
Kucuk O, Sarkar FH, Djuric Z, et al:
Effects of lycopene supplementation in patients with localized
prostate cancer. Exp Biol Med (Maywood). 227:881–885. 2002.
|
|
13
|
Liu C, Wang R, Zhang B, Hu C and Zhang H:
Protective effects of lycopene on oxidative stress, proliferation
and autophagy in iron supplementation rats. Biol Res. 46:189–200.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li H, Deng Z, Liu R, Loewen S and Tsao R:
Carotenoid compositions of coloured tomato cultivars and
contribution to antioxidant activities and protection against
H2O2-induced cell death in H9c2. Food Chem.
136:878–888. 2013. View Article : Google Scholar
|
|
15
|
Harrison D, Griendling KK, Landmesser U,
Hornig B and Drexler H: Role of oxidative stress in
atherosclerosis. Am J Cardiol. 91:7A–11A. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Klionsky DJ, Abdalla FC, Abeliovich H, et
al: Guidelines for the use and interpretation of assays for
monitoring autophagy. Autophagy. 8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Feng D, Liu L, Zhu Y and Chen Q: Molecular
signaling toward mitophagy and its physiological significance. Exp
Cell Res. 319:1697–1705. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dodson M, Darley-Usmar V and Zhang J:
Cellular metabolic and autophagic pathways: Traffic control by
redox signaling. Free Radic Biol Med. 63:207–221. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Takagi H, Matsui Y and Sadoshima J: The
role of autophagy in mediating cell survival and death during
ischemia and reperfusion in the heart. Antioxid Redox Signal.
9:1373–1381. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Thomas RL and Gustafsson AB: Mitochondrial
autophagy - an essential quality control mechanism for myocardial
homeostasis. Circ J. 77:2449–2454. 2013. View Article : Google Scholar
|
|
21
|
Kubli DA, Zhang X, Lee Y, et al: Parkin
protein deficiency exacerbates cardiac injury and reduces survival
following myocardial infarction. J Biol Chem. 288:915–926. 2013.
View Article : Google Scholar :
|
|
22
|
Nakai A, Yamaguchi O, Takeda T, et al: The
role of autophagy in cardiomyocytes in the basal state and in
response to hemodynamic stress. Nat Med. 13:619–624. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Thomas RL, Roberts DJ, Kubli DA, et al:
Loss of MCL-1 leads to impaired autophagy and rapid development of
heart failure. Genes Dev. 27:1365–1377. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nishino I, Fu J, Tanji K, et al: Primary
LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and
myopathy (Danon disease). Nature. 406:906–910. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhu H, Tannous P, Johnstone JL, et al:
Cardiac autophagy is a maladaptive response to hemodynamic stress.
J Clin Invest. 117:1782–1793. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Takagi H, Matsui Y, Hirotani S, Sakoda H,
Asano T and Sadoshima J: AMPK mediates autophagy during myocardial
ischemia in vivo. Autophagy. 3:405–407. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sciarretta S, Hariharan N, Monden Y,
Zablocki D and Sadoshima J: Is autophagy in response to ischemia
and reperfusion protective or detrimental for the heart? Pediatr
Cardiol. 32:275–281. 2011. View Article : Google Scholar
|
|
28
|
Matsui Y, Takagi H, Qu X, et al: Distinct
roles of autophagy in the heart during ischemia and reperfusion:
roles of AMP-activated protein kinase and Beclin 1 in mediating
autophagy. Circ Res. 100:914–922. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ma X, Liu H, Foyil SR, Godar RJ,
Weinheimer CJ, Hill JA and Diwan A: Impaired autophagosome
clearance contributes to cardiomyocyte death in
ischemia-reperfusion injury. Circulation. 125:3170–3181. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sala-Mercado JA, Wider J, Undyala VV,
Jahania S, et al: Profound cardioprotection with chloramphenicol
succinate in the swine model of myocardial ischemia-reperfusion
injury. Circulation. 122(Suppl 11): S179–S184. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yan W, Zhang H, Bai X, Lu Y, Dong H and
Xiong L: Autophagy activation is involved in neuroprotection
induced by hyperbaric oxygen preconditioning against focal cerebral
ischemia in rats. Brain Res. 1402:109–121. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liaw NY, Hoe LS, Sheeran FL, Peart JN,
Headrick JP, Cheung MM and Pepe S: Postnatal shifts in ischemic
tolerance and cell survival signaling in murine myocardium. Am J
Physiol Regul Integr Comp Physiol. 305:R1171–R1181. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Valentim L, Laurence KM, Townsend PA, et
al: Urocortin inhibits Beclin1-mediated autophagic cell death in
cardiac myocytes exposed to ischaemia/reperfusion injury. J Mol
Cell Cardiol. 40:846–852. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hamacher-Brady A, Brady NR and Gottlieb
RA: Enhancing macroautophagy protects against ischemia/reperfusion
injury in cardiac myocytes. J Biol Chem. 281:29776–29787. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hariharan N, Zhai P and Sadoshima J:
Oxidative stress stimulates autophagic flux during
ischemia/reperfusion. Antioxid Redox Signal. 14:2179–2190. 2011.
View Article : Google Scholar :
|
|
36
|
Sesso HD, Buring JE, Norkus EP and Gaziano
JM: Plasma lycopene, other carotenoids, and retinol and the risk of
cardiovascular disease in women. Am J Clin Nutr. 79:47–53.
2004.
|
|
37
|
Rissanen TH, Voutilainen S, Nyyssönen K,
Lakka TA, et al: Low serum lycopene concentration is associated
with an excess incidence of acute coronary events and stroke: the
Kuopio Ischaemic Heart Disease Risk Factor Study. Br J Nutr.
85:749–754. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Park S, Kim MY, Lee DH, et al: Methanolic
extract of onion (Allium cepa) attenuates ischemia/hypoxia-induced
apoptosis in cardiomyocytes via antioxidant effect. Eur J Nutr.
48:235–242. 2009. View Article : Google Scholar : PubMed/NCBI
|