Introduction
Activation, dysfunction and structural alterations
of the arterial endothelium results in the subendothelial retention
of oxidized low-density lipoprotein (ox-LDL) from the plasma,
thereby promoting the development and progression of
atherosclerosis (1). Activated
endothelial cells secrete chemokines, and subsequently express
adhesion molecules, which may facilitate the recruitment of
leukocytes, from the blood into the arterial wall (2). Of the leukocytes, monocytes are a
critical cell type that contribute to the formation of
atherosclerotic plaques. In the intima, migrated monocytes
differentiate into macrophages, which engulf ox-LDL to form
cholesterol-laden foam cells (3).
Autophagy is a conserved catabolic process in which
cytoplasmic material is delivered to the lysosomal machinery for
degradation and recycling. It is well-known that autophagy occurs
in advanced atherosclerotic plaques (4–6), and
that macrophage autophagy exerts a protective effect in advanced
atherosclerosis (7). Previous
studies have shown that ox-LDL may induce autophagy in endothelial
cells, leading to the degradation of ox-LDL through autolysosomes
(8,9). However, little is currently known
about whether ox-LDL may induce autophagy in macrophages.
Statins are inhibitors of
3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase. Statins
impede cholesterol biosynthesis, therefore reducing blood
cholesterol levels. As well as lowering lipid levels, statins have
been demonstrated to possess pleiotropic effects, including
anti-inflammatory and immunomodulatory properties (10,11).
Simvastatin is a widely used statin, that exhibits lipid and
non-lipid effects (12,13). Recently, simvastatin was shown to
enhance autophagy in coronary arterial myocytes, through inhibition
of the Rac1-mammalian target of rapamycin (mTOR) signaling pathway
(14). However, the effects of
simvastatin on macrophage autophagy remain unclear.
The present study examined whether ox-LDL may induce
autophagy in macrophages, and explored the role of simvastatin in
ox-LDL-induced macrophage autophagy and lipid accumulation.
Materials and methods
Cell culture and reagents
The J774A.1 murine macrophage cell line was
purchased from the China Center for Type Culture Collection (Wuhan,
China). The cells were maintained in RPMI-1640 medium (Thermo
Fisher Scientific, Co., Waltham, MA, USA), supplemented with 10%
fetal calf serum (Zhejiang Tianhang Biological Technology Co.,
Ltd., Hangzhou, China), at 37°C in an atmosphere containing 5%
CO2. Ox-LDL was purchased from Yiyuan Biotechnology Co.,
Ltd (Guangzhou, China). Simvastatin was purchased from Tokyo
Chemical Industry Co., Ltd (Shanghai, China). Oil Red O dye and
Total Cholesterol Detection kit were purchased from Nanjing
Jiancheng Biotechnology Institute Co., Ltd (Nanjing, China).
Lipofectamine® 2000 was obtained from Life Technologies
(Grand Island, NY, USA). Rabbit anti-microtubule-associated protein
1 light chain 3(LC3) and rabbit anti-Beclin1 antibodies were
purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA).
Horseradish peroxidase (HRP)-labeled goat anti-rabbit
Immunoglobulin G was purchased from Beijing ZSGB-Biotechnology, Co.
(Beijing, China). The Bicinchoninic Acid (BCL) Protein Assay kit
was obtained from Beijing YPH-Bio Company (Beijing, China), and the
Enhanced Chemiluminescence (ECL) Detection system was purchased
from Pierce Biotechnology, Inc. (Rockville, MD, USA).
Establishment and detection of macrophage
foam cell model
The J774A.1 cells were seeded into 6-well plates
(5×105 cells per well) and grown to 50–60% confluency. A
total of 0, 25, 50 or 100 μg/ml ox-LDL was added to the cell
supernatants, and the cells were incubated for 24 h. The cells were
washed with phosphate-buffered saline and fixed with formaldehyde.
The fixed cells were stained with oil Red O dye, for 30 min at
37°C. The status of intracellular lipid accumulation was detected
by microscopy (Olympus CKX41; Olympus, Tokyo, Japan), in order to
assess the formation of macrophage foam cells.
Western blotting
The J774A.1 cells were plated in 6-well plates and
grown to 50–60% confluency. Following stimulation with ox-LDL
(or/and simvastatin at 0.5 and 1.0 μM), the cells were harvested
and lysed with RIPA lysis buffer on ice. The cell lysates were
centrifuged at 13,000 g for 30 min at 4°C. The protein
concentrations of the supernatants were determined using the BCA
kit. Equal quantities of protein (30 μg) from the homogenized
samples were separated by 15% SDS-PAGE, and transferred to
polyvinylidene fluoride membranes (Millipore, Boston, MA, USA). The
membranes were blocked with tris-buffered saline-Tween®
(TBST), containing 5% non-fat milk powder, for 2 h at room
temperature. The membranes were then incubated with polyclonal
rabbit anti-microtubule-associated protein 1 light chain 3(LC3;
1:1,000) and polyclonal rabbit anti-Beclin1 (1:1,000) primary
antibodies at 4°C overnight, washed with TBST, and incubated for 1
h with HRP-conjugated goat anti-rabbit antibody (1:10,000
dilution), at room temperature. β-actin was used as a control.
Immunoreactive bands were visualized using the ECL detection system
and images were captured using an automatic digital gel image
analysis system (4500SF; Tanon Science & Technology Co., Ltd.,
Shanghai, China).
Confocal laser scanning microscopy
Green fluorescent protein (GFP)-LC3 plasmids were
previously prepared in our laboratory (15) and were transfected into the J774A.1
cells, using Lipofectamine® 2000 according to the
manufacturer’s instructions. The cells were treated with ox-LDL
(and/or simvastatin) 24 h post-transfection, and incubated for a
further 24 h. All GFP-LC3 plasmid-transfected cells with or without
ox-LDL (and/or simvastatin) treatment were then fixed using Immunol
Staining Fix Solution (Beyotime Institute of Biotechnology, Haimen,
China) and GFP-LC3 fluorescence was detected by confocal laser
scanning microscopy (Leica TCS SP5; Leica, Mannheim, Germany).
Measurement of intracellular cholesterol
using the CHOD-PAP method
Following stimulation with ox-LDL (or/and
simvastatin), the cells were harvested and sonicated on ice for a
total of 5 sec, with 15 sec pauses between bursts using a sonicator
(Ningbo Scientz Biotechnology Co., Ltd., Ningbo, China). Following
sonication for 10 min, the cells were centrifuged at 8,000 g for 10
min at 4°C. The supernatants were then used for measurement of
intracellular cholesterol. Total cholesterol levels in the cells
were determined by the CHOD-PAP method, using the Total Cholesterol
Detection kit (Nanjing Jiancheng Biotechnology Institute Co.,
Ltd.), according to the manufacturer’s instructions.
Statistical analysis
The data are represented as the mean ± standard
deviation, and were statistically analyzed using SPSS 13.0 software
(SPSS Inc., Chicago, IL, USA). Statistical analysis was performed
using Student’s t-test to compare the differences between two
groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
Ox-LDL induces lipid accumulation in
macrophages
To investigate the possible roles of simvastatin on
ox-LDL-induced macrophage autophagy and lipid accumulation, a
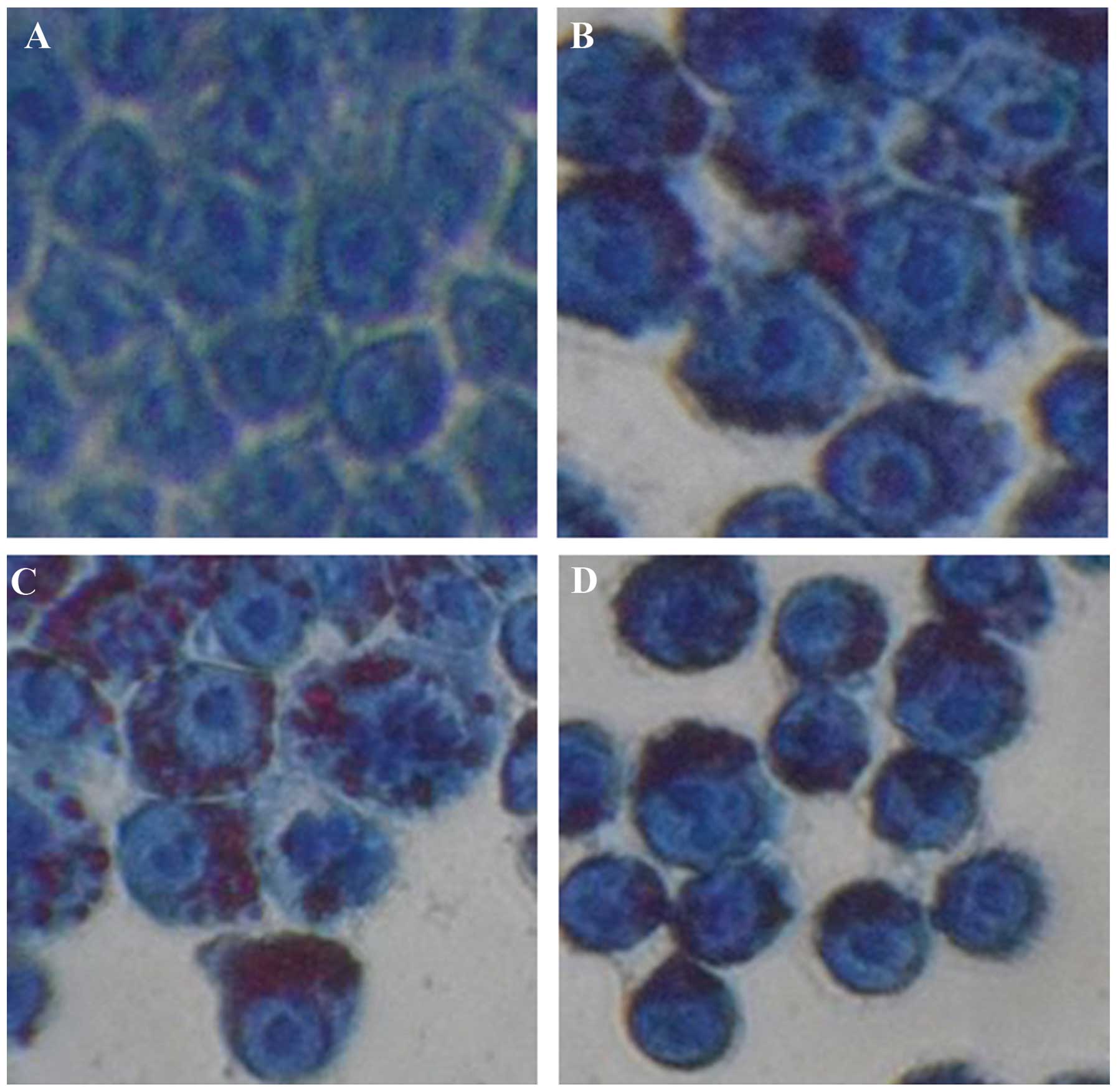
macrophage foam cell model was generated. Groups A, B, C and D of
the J774A.1 cells were treated with 0, 25, 50 and 100 μg/ml ox-LDL
respectively, for 24 h, and the status of lipid accumulation was
determined by oil red O staining. No obvious lipid accumulation was
observed in the control group (Fig.
1A). Conversely, lipid accumulation was observed in all of the
groups treated with ox-LDL (Fig.
1B–D). Lipid accumulation was more visible in groups C and D,
as compared with group B, implying the existence of a
dose-dependent effect. All of the cells treated with ox-LDL had
similar morphological characteristics to foam cells, suggesting the
successful establishment of a macrophage foam cell model.
Thereafter, 50 μg/ml ox-LDL was selected to treat cells in further
experiments.
Ox-LDL induces the transformation of LC3
I to LC3 II in macrophages
Ox-LDLs have previously been shown to trigger
autophagy in endothelial cells (8,9,16).
The present study examined whether ox-LDL induced autophagy in
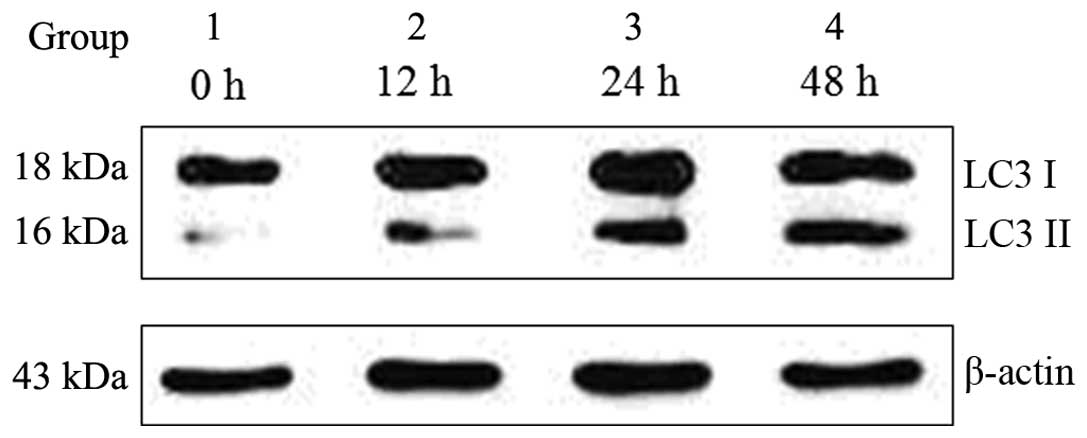
macrophages. Ox-LDL (50 μg/ml) induced autophagy in the J774A.1
macrophage cell line by converting LC3 I to LC3 II, which is a
well-known autophagy marker. The transformation of LC3 I to LC3 II
increased gradually, in a time-dependent manner following treatment
with 50 μg/ml ox-LDL, as compared with the control group (Fig. 2).
Simvastatin enhances ox-LDL-induced
macrophage autophagy
Simvastatin was previously shown to enhance
autophagy in osteoblastic cells and coronary arterial myocytes
(14,17). The present study determined whether
simvastatin elevated ox-LDL-induced autophagy in macrophages.
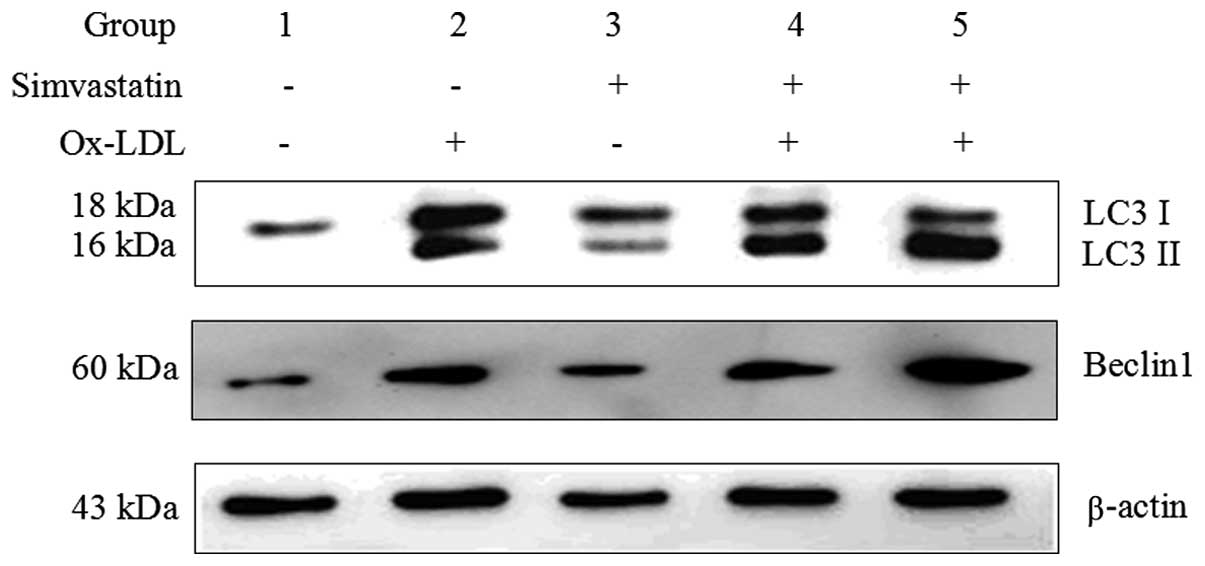
Concordant with the results presented in Fig. 2, the protein expression levels of
the autophagy marker LC3 II were induced in group 2, following
stimulation with 50 μg/ml ox-LDL for 24 h (Fig. 3). Notably, treatment with
simvastatin markedly promoted the conversion of LC3 I to LC3 II
(group 4 and 5, as compared with group 2). Treatment with 1.0 μM
simvastatin enhanced ox-LDL-induced LC3 II expression more than
treatment with 0.5 μM simvastatin, suggesting that simvastatin
affects ox-LDL-induced autophagy in a dose-dependent manner
(Fig. 3). Simvastatin (0.5 and 1.0
μM) also increased the expression levels of another ox-LDL-induced
autophagy marker, Beclin1 (Fig.
3).
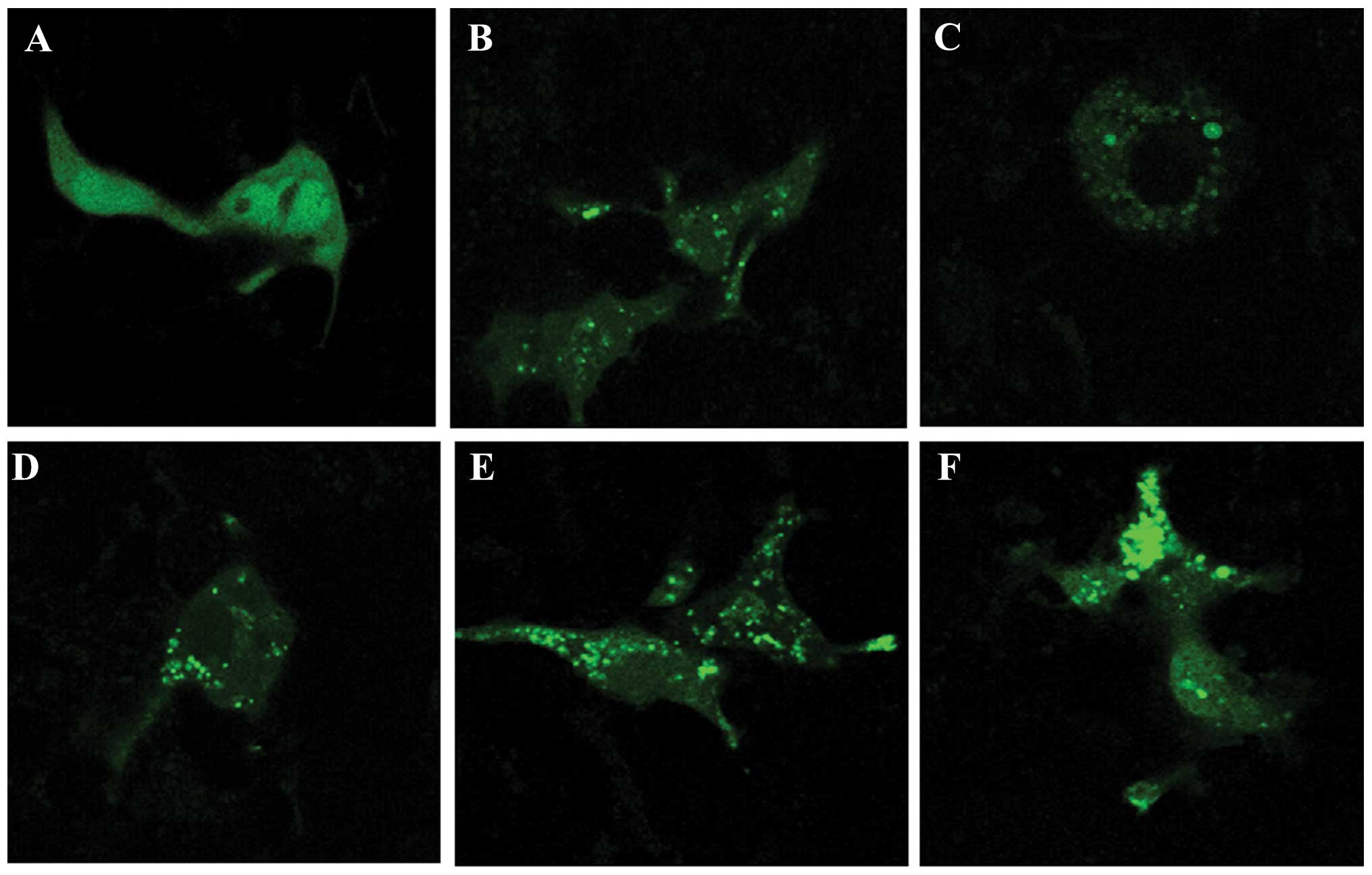
The GFP-LC3 plasmid was used to detect autophagosome
formation. Stimulation with ox-LDL led to the redistribution of
GFP-LC3, from diffusion distribution to puncta in the J774A.1
cells, as determined by confocal laser scanning microscopy
(Fig. 4A and B). Treatment with
simvastatin (0.5 and 1.0 μM) alone resulted in the formation of
GFP-LC3 fluorescent puncta in the J774A.1 cells (Fig. 4C and D). Furthermore, treatment
with simvastatin markedly promoted the ox-LDL-induced formation of
GFP-LC3 puncta (Fig. 4E and F).
Treatment with 1.0 μM simvastatin enhanced the formation of
ox-LDL-induced GFP-LC3 puncta more so than 0.5 μM simvastatin
(Fig. 4E and F). These results
indicate that simvastatin may enhance ox-LDL-induced macrophage
autophagy.
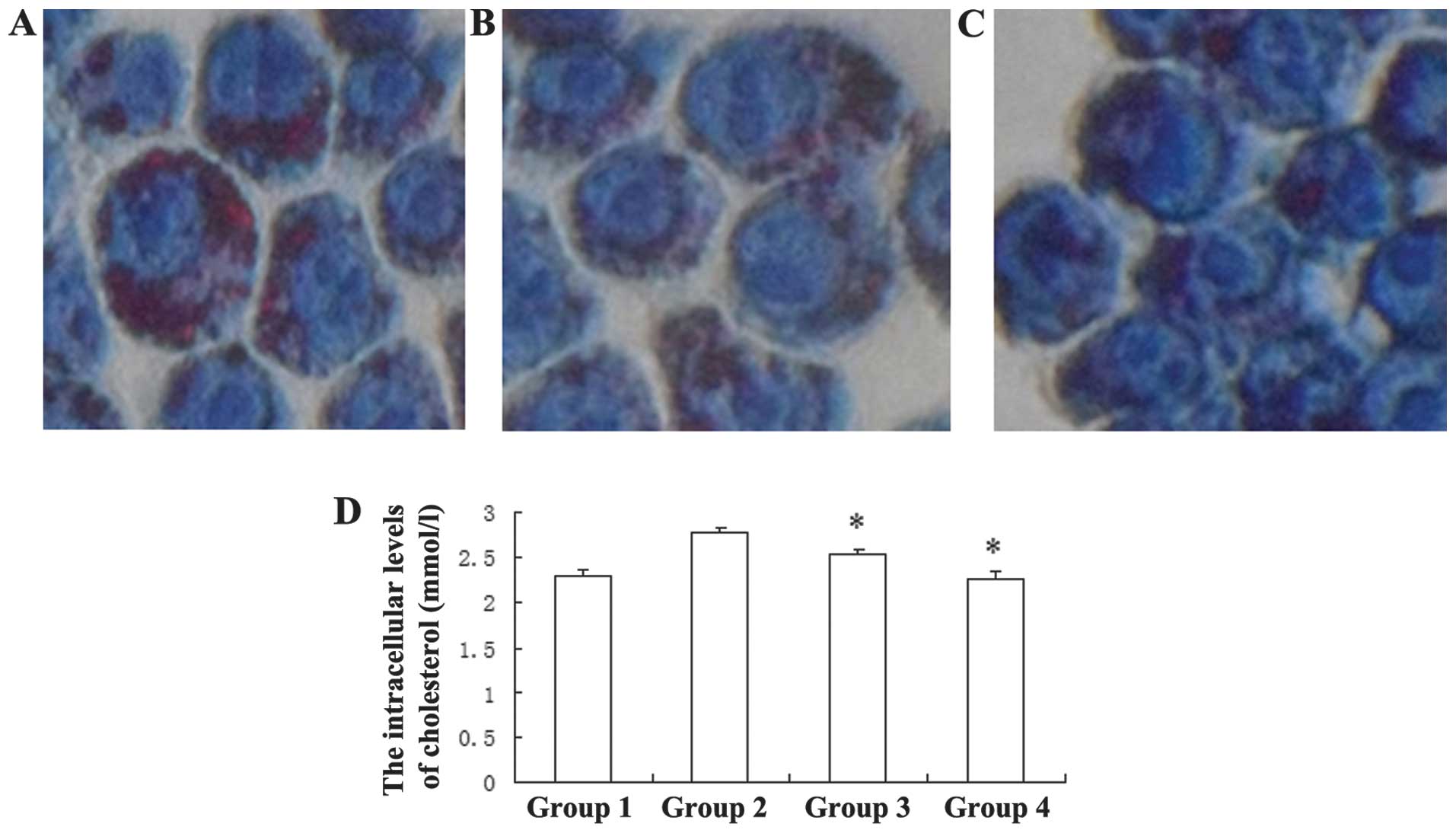
Simvastatin inhibits ox-LDL-induced
cholesterol accumulation in macrophages
As a HMG-CoA reductase inhibitor, simvastatin is a
well-known drug used to ameliorate atherosclerotic diseases, by
lowering plasma cholesterol. The present study determined whether
simvastatin may inhibit ox-LDL-induced cholesterol accumulation in
J774A.1 cells. The number of lipid droplets, the existing form of
cholesterol in cells, decreased following incubation with
simvastatin (Fig. 5A–C). The
CHOD-PAP assay indicated that treatment with 0.5 and 1.0 μM
simvastatin led to a significant reduction in the intracellular
total cholesterol levels, from 2.774±0.059 mmol/l, to 2.53±0.058
mmol/l and 2.27±0.085 mmol/l, respectively (Fig. 5D). These results suggest that
simvastatin may inhibit ox-LDL-induced cholesterol accumulation in
macrophages.
Discussion
Ox-LDL has a pivotal role in atherosclerosis
initiation and progression. Ox-LDL may be recognized and engulfed
by macrophages through scavenger receptors, leading to the
formation of foam cells, which are the prominent feature of
atherosclerotic lesions (18,19).
As predicted, the results of the present study demonstrated that
ox-LDL was capable of inducing lipid accumulation in J774A.1 cells,
indicating the formation of foam cells. Previous studies have shown
that ox-LDL may induce autophagy in endothelial cells (8,9,16,20).
However, it is relatively difficult to observe autophagy in
macrophages, since they are phagocytic and the cytoplasmic vacuoles
render it difficult to distinguish between autophagocytosis and
heterophagocytosis (4). Ouimet and
Marcel (21) reported that
acetylated-LDL elevated macrophage intracellular cholesterol
levels, and were delivered to the lysosomes by autophagy (21). Concordant with these results, the
present study demonstrated that ox-LDL induced autophagy in
macrophages, by increasing the expression of LC3 II and Beclin1,
the classic autophagy markers.
As a powerful lipid-lowering drug, simvastatin is
widely used for the treatment of atherosclerosis. Accumulating
evidence has shown that simvastatin also has various other effects.
The present study reported that, besides decreasing ox-LDL-induced
cholesterol accumulation, simvastatin intensified ox-LDL-induced
macrophage autophagy. However, it remains noteworthy that drugs may
directly induce macrophage autophagy in atherosclerosis, and
certain drug-induced macrophage autophagy promotes a stable plaque
phenotype (22). The results of
the present study showed that simvastatin may directly induce
macrophage autophagy, albeit only marginally. Whether this effect
may have a role in plaque progression requires further
investigation.
In mammalian cells there are two types of autophagy:
mTOR-dependent and mTOR-independent autophagy (22). Wei et al (14) demonstrated that simvastatin induced
autophagy through inhibition of Rac1-mTOR signaling in coronary
arterial myocytes. However, it remains unknown whether simvastatin
promotes ox-LDL-induced macrophage autophagy in the same way.
Further studies are required to explore whether ox-LDL-induced
macrophage autophagy is mTOR-dependent or not. In addition, the
results of the present study were obtained from in vitro
experiments. An animal model of atherosclerosis may be useful to
confirm these findings.
In conclusion, the results of the present study
suggest that ox-LDL is capable of inducing autophagy in
macrophages, and simvastatin may enhance ox-LDL-induced macrophage
autophagy and attenuate lipid accumulation. These results imply
that macrophage autophagy in atherosclerosis may be the potential
target of simvastatin for plaque stabilization.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China Grants (grant no.
30972791, to Baojun Huang) and the Research Fund of Anhui Medical
University (grant no. 0116025101, to Songcheng Ying).
References
|
1
|
Legein B, Temmerman L, Biessen EA and
Lutgens E: Inflammation and immune system interactions in
atherosclerosis. Cell Mol Life Sci. 70:3847–3869. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cybulsky MI, Won D and Haidari M:
Leukocyte recruitment to atherosclerotic lesions. Can J Cardiol.
20:24B–28B. 2004.PubMed/NCBI
|
|
3
|
Moore KJ, Sheedy FJ and Fisher EA:
Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol.
13:709–721. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Martinet W and De Meyer GR: Autophagy in
atherosclerosis. Curr Atheroscler Rep. 10:216–223. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Martinet W and De Meyer GR: Autophagy in
atherosclerosis: a cell survival and death phenomenon with
therapeutic potential. Circ Res. 104:304–317. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schrijvers DM, De Meyer GR and Martinet W:
Autophagy in atherosclerosis: a potential drug target for plaque
stabilization. Arterioscler Thromb Vasc Biol. 31:2787–2791. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liao X, Sluimer JC, Wang Y, Subramanian M,
Brown K, Pattison JS, Robbins J, Martinez J and Tabas I: Macrophage
autophagy plays a protective role in advanced atherosclerosis. Cell
Metab. 15:545–553. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang YL, Cao YJ, Zhang X, Liu HH, Tong T,
Xiao GD, Yang YP and Liu CF: The autophagy-lysosome pathway: a
novel mechanism involved in the processing of oxidized LDL in human
vascular endothelial cells. Biochem Biophys Res Commun.
394:377–382. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Muller C, Salvayre R, Nègre-Salvayre A and
Vindis C: HDLs inhibit endoplasmic reticulum stress and autophagic
response induced by oxidized LDLs. Cell Death Differ. 18:817–828.
2011. View Article : Google Scholar :
|
|
10
|
Almuti K, Rimawi R, Spevack D and Ostfeld
RJ: Effects of statins beyond lipid lowering: potential for
clinical benefits. Int J Cardiol. 109:7–15. 2006. View Article : Google Scholar
|
|
11
|
Bu DX, Griffin G and Lichtman AH:
Mechanisms for the anti-inflammatory effects of statins. Curr Opin
Lipidol. 22:165–170. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wierzbicki AS, Poston R and Ferro A: The
lipid and non-lipid effects of statins. Pharmacol Ther. 99:95–112.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Marzilli M: Pleiotropic effects of
statins: evidence for benefits beyond LDL-cholesterol lowering. Am
J Cardiovasc Drugs. 10(Suppl 1): 3–9. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wei YM, Li X, Xu M, Abais JM, Chen Y,
Riebling CR, Boini KM, Li PL and Zhang Y: Enhancement of autophagy
by simvastatin through inhibition of Rac1-mTOR signaling pathway in
coronary arterial myocytes. Cell Physiol Biochem. 31:925–937. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang M, Huang BJ, Hu MC, Chen YW, Song W,
Huang DK, Li Q and Hu CS: Lipopolysaccharide induces formation of
autophagy-related LC3B-GFP fluorescent aggregates in the RAW264.7
macropahge cells. J Anhui Medical Uni. 47:357–361. 2012.(In
Chinese).
|
|
16
|
Nowicki M, Zabirnyk O, Duerrschmidt N,
Borlak J and Spanel-Borowski K: No upregulation of lectin-like
oxidized low-density lipoprotein receptor-1 in serum-deprived
EA.hy926 endothelial cells under oxLDL exposure, but increase in
autophagy. Eur J Cell Biol. 86:605–616. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lai EH, Hong CY, Kok SH, Hou KL, Chao LH,
Lin LD, Chen MH, Wu PH and Lin SK: Simvastatin alleviates the
progression of periapical lesions by modulating autophagy and
apoptosis in osteoblasts. J Endod. 38:757–763. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Maiolino G, Rossitto G, Caielli P, Bisogni
V, Rossi GP and Calò LA: The role of oxidized low-density
lipoproteins in atherosclerosis: the myths and the facts. Mediators
Inflamm. 2013:7146532013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mitra S, Goyal T and Mehta JL: Oxidized
LDL, LOX-1 and atherosclerosis. Cardiovasc Drugs Ther. 25:419–429.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ding Z, Wang X, Khaidakov M, Liu S, Dai Y
and Mehta JL: Degradation of heparan sulfate proteoglycans enhances
oxidized-LDL-mediated autophagy and apoptosis in human endothelial
cells. Biochem Biophys Res Commun. 426:106–111. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ouimet M and Marcel YL: Regulation of
lipid droplet cholesterol efflux from macrophage foam cells.
Arterioscler Thromb Vasc Biol. 32:575–581. 2012. View Article : Google Scholar
|
|
22
|
Martinet W, De Meyer I, Verheye S,
Schrijvers DM, Timmermans JP and De Meyer GR: Drug-induced
macrophage autophagy in atherosclerosis: for better or worse? Basic
Res Cardiol. 108:3212013. View Article : Google Scholar
|