Introduction
Gliomas are the most common type of malignant
primary intracranial neoplasm and are associated with high
mortality and morbidity (1).
Despite dramatic advances in surgical intervention and chemotherapy
over the past decades, the prognosis for patients with malignant
glial tumors remains poor (2).
Glioblastoma multiforme is the most common and aggressive subtype
of malignant glioma, with an average survival time of less than one
year following diagnosis (2).
Accordingly, there is an urgent requirement to develop novel and
effective therapeutic strategies for this disease.
Temozolomide (TMZ) is a standard chemotherapeutic
agent for gliomas following surgical resection and radiotherapy
(3). Functioning as an alkylating
agent, TMZ induces DNA damage via the attachment of a methyl group
to guanine bases, resulting in disrupted gene expression and cell
growth arrest. However, not all patients exhibit a sensitive
response to TMZ, and acquired resistance to the drug following
treatment is frequently observed (4,5).
Accordingly, an understanding of the molecular mechanisms
underlying TMZ resistance is essential for the optimization of
existing therapeutic strategies and the development of new ones
(6).
Estrogens are steroid hormones that play a
significant role in regulating the development and differentiation
of the central nervous system (7,8).
There are two main types of cognate receptors of estrogens, ERα and
ERβ, which selectively bind to different ligands and mediate the
expression of different downstream genes and signaling cascades
(7,9,10).
ERα has been shown to enhance the proliferation of cancer cells
(10–12), while previous studies support the
role of ERβ as a potential tumor suppressor (7,10,13,14).
Loss of ERβ expression has been repeatedly observed in high-grade
glioma tumors and is associated with poor clinical outcome
(10,14,15).
A previous study also revealed that overexpression of ERβ reduced
cell proliferation in colon and breast cancer cells while knockdown
of ERβ exhibited the opposite effect (13). Moreover, it has been reported that
treatment with ERβ-specific agonists results in proliferation
arrest and/or induced cell death in a wide range of cancer cells
(10,11). Accordingly, the expression level of
ERβ may be a key determinant of cellular responses to antitumor
drugs, while ERβ-specific agonists may serve as a potential
treatment for gliomas.
Liquiritigenin (Liq) is a herb-derived, highly
selective ERβ agonist (16). It
activates multiple regulatory elements and downstream target genes
of ERβ with high specificity, and exhibits various
anti-inflammatory (17,18) and antitumor effects (19,20).
It has been shown that Liq inhibits the production of inducible
nitric oxide synthase and the release of proinflammatory cytokines
in macrophages (17). There is
also evidence that Liq suppresses cell proliferation in a dose- and
time-dependent manner (20).
Pharmacokinetic studies demonstrate that Liq exhibits strong
intestinal absorption and blood-brain barrier permeability
(20). These characteristics all
support the therapeutic function of Liq in gliomas. However,
besides its well-established role as a tumor suppressor, little is
known about the function of ERβ in regulating the chemotherapeutic
response of gliomas. To investigate the therapeutic advantages of
the ERβ agonist, we considered whether it was possible to modulate
ERβ signaling using Liq and enhance cellular sensitivity to TMZ.
These findings provide implications for the molecular mechanisms of
TMZ resistance and have immediate clinical utility in glioma
treatment.
Materials and methods
Cell lines and reagents
U138 human glioma cells were obtained from the
American Type Culture Collection (Manassas, VA, USA) and cultured
in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal
bovine serum (Hyclone Laboratories, Logan, UT, USA) and 1%
penicillin-streptomycin (Sigma, St. Louis, MO, USA). Liq was
purchased from Biopurify Phytochemicals (Chengdu, China). AKT
antibody (no. 9272), phospho-AKT antibody (no. 9271) and
phospho-p70S6K antibody (no. 9205) were purchased from Cell
Signaling Technology, Inc. (Danvers, MA. USA). P70S6K antibody
(ab9366) was purchased from Abcam (Cambridge, UK). ERβ antibody
(sc-8974), ERβ shRNA (h) lentiviral particles (sc-35325-V) and
control shRNA lentiviral particles-A (sc-108080) were purchased
from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). All
antibodies are rabbit anti-human polyclonal antibodies.
Reverse-transcription polymerase chain
reaction (PCR) and real-time PCR
TRIzol reagent (Invitrogen Life Technologies,
Carlsbad, CA, USA) was used to extract total RNA following the
manufacturer’s instructions. RNA was quality checked and
quantitated using Nanodrop (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). Total RNA (500 μM) was used for cDNA synthesis
with poly T primer using the Superscript III reagent kit
(Invitrogen Life Technologies). Real-time PCR was performed using a
7300 real-time PCR system (Applied Biosystems, Life Technologies,
Paisley, UK) using SYBR-Green PCR master mix (Bio-Rad, Hercules,
CA, USA). Data are expressed as the mean ± standard deviation of
three replicates. The PCR primers used for ERβ were:
5′-GGCAGAGGACAGTAAAAGCA-3′ (forward) and 5′-GGACCACACAGCAGAAAGAT-3′
(reverse). For actin, they were: 5′-CCAACACAGTGCTGTCTGG-3′
(forward) and 5′-TGCTGATCCACATCTGCTG-3′ (reverse).
Western blotting
Cells were washed twice with ice-cold
phosphate-buffered saline and lysed in protein gel buffer protein
gel buffer (PGB; 60 mM Tris-HCl,10% SDS and 10% glycerol) with 1 mM
phenylmethanesulfonylfluoride for 20 min on ice. PGB buffer
contains 60 mM Tris-HCl, 10% SDS and 10% glycerol. Cell lysates
were centrifuged at 14,000 × g for 10 min at 4°C, and the
supernatants were collected. Protein concentration was quantitated
using Nanodrop spectrophotometer nd-1000 (Thermo Fisher Scientific,
Inc.) and 10 μg protein was loaded on 12% SDS-PAGE gels and
transferred onto membranes (Millipore, Billerica, MA, USA).
Membranes were blocked in TBST with 5% non-fat milk at room
temperature for 1 h, and incubated with primary antibodies at 4°C
overnight. The next day, membranes were washed three times for 5
min with TBST, incubated with horseradish peroxidase-conjugated
secondary antibodies for 1 h at room temperature and visualized
using enhanced chemiluminescence (Millipore). Primary antibodies
were diluted at a concentration of 1:1,000, with the exception of
anti-AKT (1:2,000) and anti-ERβ (1:500).
shRNA lentiviral particle
transduction
U87 cells were cultured at a density of
2×104 cells per well in 12-well plates and cultured for
24 h prior to infection. The cells were approximately 50% confluent
when they were infected with ERβ-specific shRNA lentiviral
particles or control shRNA lentiviral particles in a mixture of
complete medium containing 5 μg/ml polybrene (Santa Cruz
Biotechnology, Inc.). Following incubation for 24 h, the culture
medium was replaced with fresh complete medium and incubation was
continued for 24 h. Next, the cells were divided into three groups
and transferred to new plates for subsequent assays. Knockdown
efficiency was accessed using western blotting.
Cell viability assay
The effects of different agent combinations on cell
viability were analyzed using the Cell Counting Kit 8 (CCK-8) cell
viability assay (Dojindo, Kunamoto, Japan). Briefly, cells were
suspended in 96-well plates at a density of 104 cells
per well. Following incubation for 24 h, various agents, alone or
in combination with others as indicated in the manuscript, were
added to the cells. Following incubation for 72 h, 10 μl CCK-8
reagent was added to each well and incubated at 37°C for 2 h.
Absorbance was accessed at a wavelength of 450–630 nm using a
multi-mode microplate reader (BioTek Instruments, Inc., Winooski,
VT, USA). Assays were performed in triplicate and cell viability
was calculated as a percentage of the control. The median
inhibitory concentration (IC50) of TMZ was calculated
using SPSS software 17.0 (SPSS, Inc., Chicago, IL, USA).
Results
Liq enhances expression of ERβ and
sensitizes glioma cells to TMZ treatment
The TMZ-resistant U138 glioma cell line (21) was used as a model to investigate
the function of Liq in modulating the cellular response to TMZ
treatment. U138 cells were treated with seven different titrations
of Liq, and cell viability was determined using the CCK-8 assay
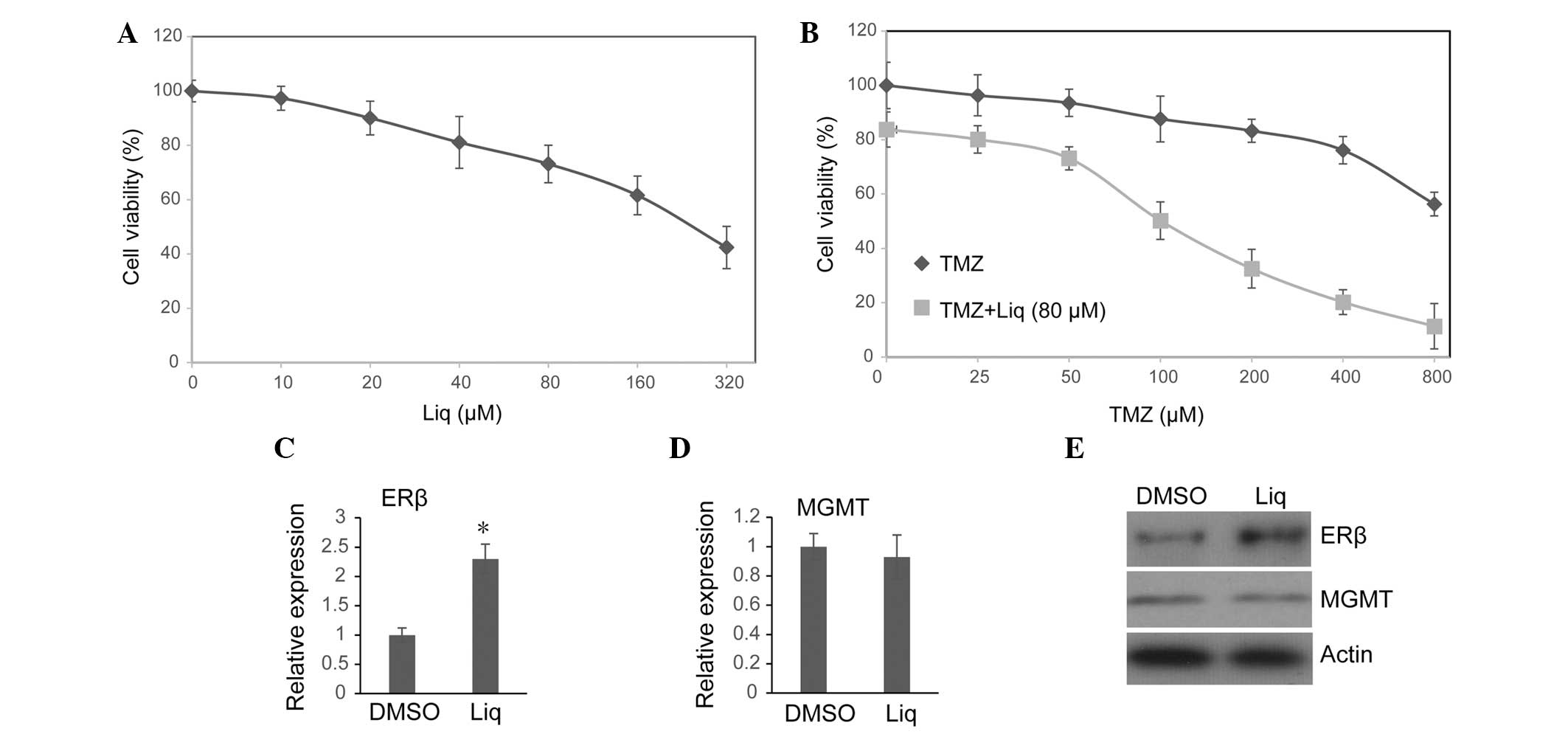
after 72 h. In agreement with a previous study (20), it was observed that Liq induced
U138 cell death in a dose-dependent manner (Fig. 1A). As 80 μM Liq was sufficient to
activate ERβ without causing severe cell death in U138 cells, this
concentration of Liq was used in all subsequent assays. The effect
of treatment with TMZ alone or in combination with Liq on cell
viability was then investigated. Consistent with previous results
(21), a clear inhibitory effect
on U138 cell viability was observed only when a high dose of TMZ
was used (Fig. 1B). By contrast,
the combined treatment of TMZ and Liq synergistically inhibited the
proliferation of U138 cells even when a low dose of TMZ was used
(Fig. 1B). For cells treated with
Liq and various doses of TMZ, we controlled for the influence of
Liq on cell viability by normalizing viabilities of different
groups to the group treated with 80 μM Liq alone (TMZ=0 μM), and
calculated a corrected 72 h IC50 for TMZ. Combined
treatment with Liq significantly increased U138 susceptibility to
TMZ (untreated IC50=973.56 μM vs. treated
IC50=188.45 μM). Since the combination of a low dose of
Liq (80 μM) and TMZ (100 μM) could readily inhibit cell growth by
~50% (Fig. 1D), this therapy
option may be well tolerated with fewer side effects. Notably,
using real-time PCR, it was observed that transcription activity of
ERβ was notably increased following treatment with 80 μM Liq for 72
h (P=0.006, Student’s t-test; Fig.
1C). Western blotting revealed that the protein level of ERβ
was also enhanced following exposure to Liq (Fig. 1E). This is in line with a previous
study which demonstrated that the expression level of ERβ is
self-regulated by its own ligands (22). By contrast, we noted that Liq
treatment had no influence on either the transcription or protein
expression of O6-methylguanine DNA
methyltransferase (MGMT) (Fig. 1D and
E), which plays a significant role in repairing TMZ-induced DNA
damage and regulating chemoresistance to alkylating agents
(4,5,21).
We therefore suggest that Liq may enhance the TMZ sensitivity of
U138 cells through MGMT-independent mechanisms.
Liq treatment results in inhibition of
PI3K/AKT/mTOR pathway
The PI3K/AKT/mTOR pathway plays a significant role
in regulating cellular processes that are critical for both normal
development and tumorigenesis, including proliferation, growth,
survival and mobility (23–25).
There is also growing evidence to suggest that the PI3K/AKT/mTOR
pathway plays a crucial role in regulating chemotherapy resistance
in various tumor cells (23,26).
In addition, a previous study suggested that the overexpression of
ERβ is associated with inhibition of the PI3K/AKT/mTOR pathway
(27,28). We therefore considered whether Liq
modulates TMZ sensitivity by regulating the PI3K/AKT/mTOR pathway.
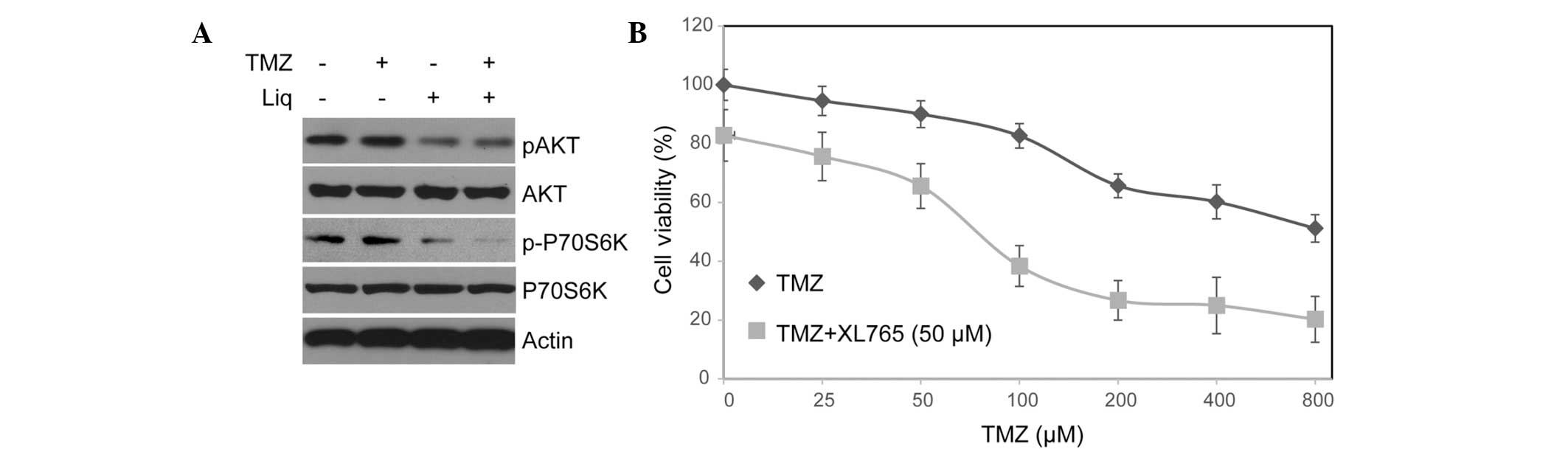
After treating cells with TMZ or Liq alone and in combination, we
investigated changes in the protein expression and phosphorylation
of AKT and P70S6K, which are central components of this pathway,
using western blotting. Notably, treatment with Liq, but not TMZ,
resulted in a significant decrease in AKT and p70S6K
phosphorylation (Fig. 2A). The
function of the PI3K/AKT/mTOR pathway in regulating TMZ sensitivity
of U138 cells was further investigated using a PI3K/mTOR dual
inhibitor, XL765 (29). Notably,
U138 cells treated with XL765 became more sensitive to TMZ even
after controlling for the influence of XL765 (untreated
IC50=897.25 μM vs. treated IC50=101.36 μM).
These results support the role of PI3K/AKT/mTOR as a key mediator
of TMZ resistance in U138 cells.
Therapeutic efficacy of Liq is
ERβ-dependent
Although it has been suggested that Liq targets ERβ
with high specificity, it may still exhibit certain off-target
activities and induce complex cellular effects. To explore whether
the function of Liq in regulating TMZ resistance is ERβ-dependent,
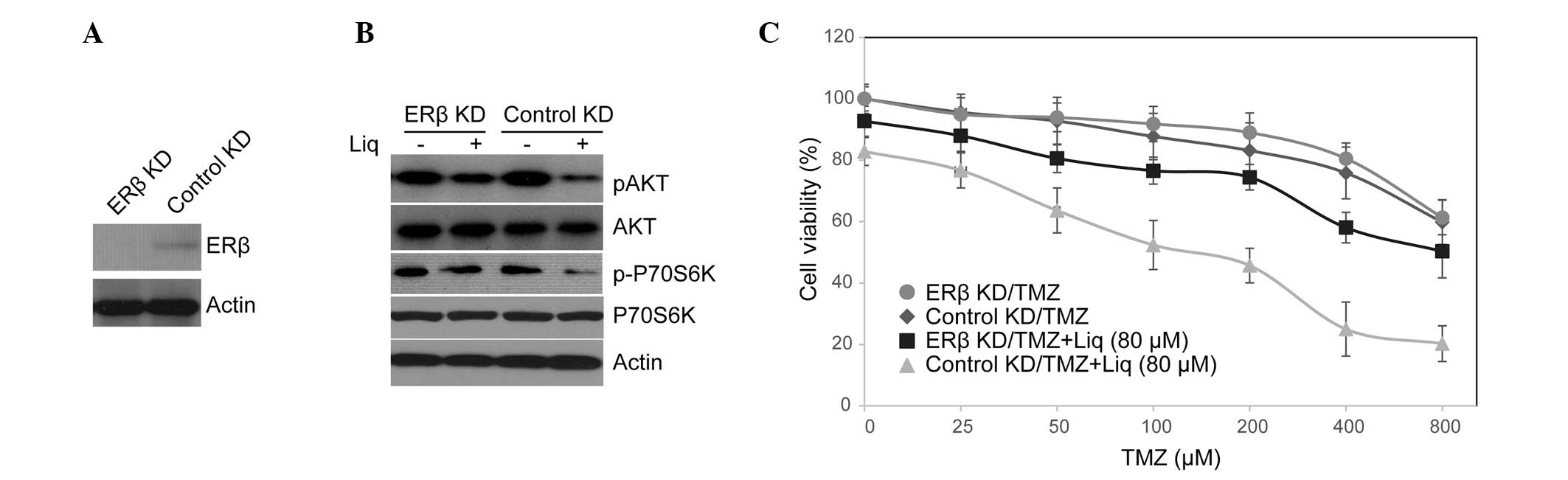
we knocked down ERβ in U138 cells with lentiviral shRNA, and tested
the activity of the PI3K/AKT/mTOR pathway as well as TMZ
sensitivity in these cells. As shown in Fig. 3A, the expression of ERβ was
markedly depressed in knockdown cells. Notably, ERβ knockdown
rescued phosphorylation of AKT and P70S6K, that were otherwise
missing in Liq-treated cells (Fig.
3B). Consistent with this observation, upon treatment with
combined TMZ and Liq, ERβ knockdown resulted in a concomitant
increase in cell proliferation compared with control cells
(knockdown IC50=597.35 μM vs. control
IC50=204.28 μM; Fig.
3C). This was not likely to be caused by ERβ knockdown per
se as knockdown cells exhibited no increase in cell viability
compared with control cells when they were treated with TMZ alone
(knockdown IC50=967.35 μM vs. control
IC50=934.28 μM). We thus conclude that the therapeutic
efficacy of Liq in conferring TMZ sensitivity is dependent on ERβ
function.
Liq function is counterbalanced by
IGF-1-induced activation of PI3K/AKT/mTOR
Through activation of ERβ, Liq may regulate the
activities of several signaling cascades that mediate TMZ
resistance in U138 cells. This indicates that the PI3K/AKT/mTOR
pathway may not necessarily be the main target of Liq/ERβ that
conferred protection against TMZ-induced cell growth inhibition. To
further evaluate the function of PI3K/AKT/mTOR signaling in TMZ
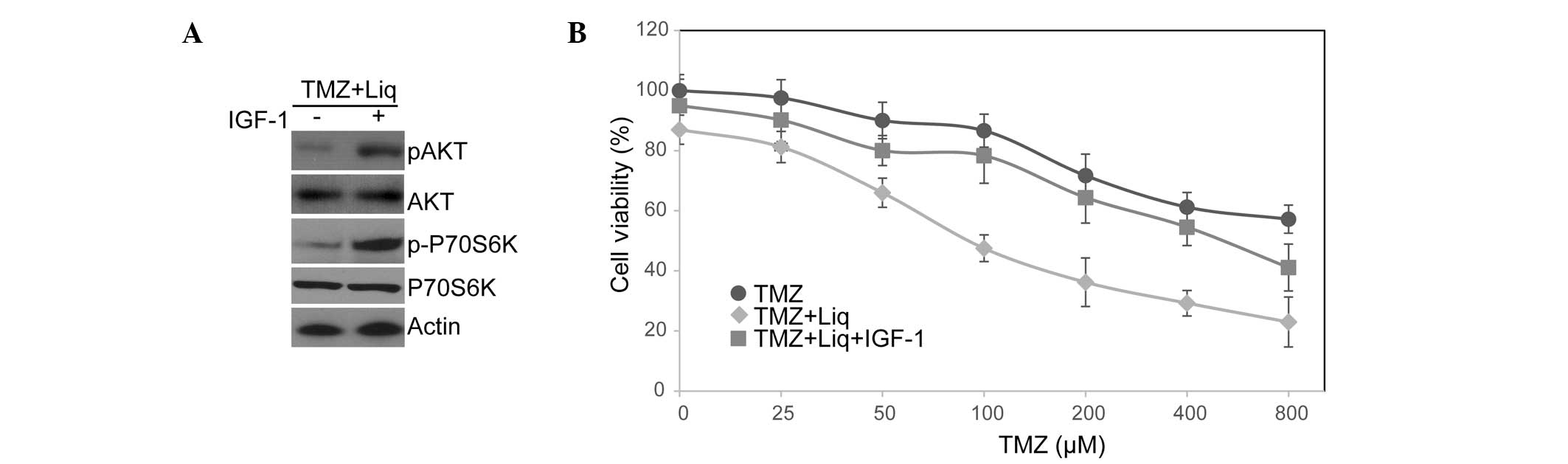
resistance, we treated U138 cells with IGF-1 to activate this
pathway and examined the cellular response to TMZ/Liq treatment.
The binding of IGF-1 to its receptor causes receptor
autophosphorylation and the activation of multiple signaling
pathways including PI3K/AKT/mTOR signaling (30). As expected, IGF-1 treatment
resulted in the superactivation of PI3K/AKT/mTOR signaling and
hyperphosphorylation of AKT and p70S6K (Fig. 4A). Moreover, IGF-1 treatment
largely counterbalanced the function of Liq in sensitizing U138
cells to TMZ (treated IC50=568.35 μM vs. untreated
IC50=174.28 μM; Fig.
4B). This is consistent with the PI3K/AKT/mTOR pathway being
significant in mediating chemoresistance (23). We thus conclude that Liq treatment
sensitizes U138 cells to TMZ through inhibition of the
PI3K/AKT/mTOR pathway.
Discussion
TMZ has emerged as a promising chemotherapy for
glioblastoma; however, the clinical outcome of TMZ treatment is not
always satisfactory due to the intrinsic or acquired drug
resistance (2). It is well
established that MGMT is one of the most significant DNA repair
enzymes that targets TMZ-induced DNA damage (4). Previous studies suggest that the
promoter methylation status and expression level of MGMT are
associated with the susceptibility of tumor cells to TMZ (4,5).
Besides MGMT-dependent mechanisms, however, other mechanisms are
also considered to be critical in regulating TMZ sensitivity
(25). Accordingly, other
pharmacological agents, through MGMT-dependent or -independent
mechanisms, may readily increase the therapeutic efficacy of TMZ
and increase patients’ survival.
In the current study, a mechanistic link was
identified between the ERβ agonist and chemoresistance to TMZ
treatment. Notably, it was revealed that Liq enhanced the TMZ
sensitivity of U138 cells without changing the expression level of
MGMT. The results provided evidence that Liq enhanced TMZ
sensitivity through inhibition of the PI3K/AKT/mTOR pathway. We
also suggest that Liq function is dependent on ERβ and may be
counterbalanced by the PI3K/AKT/mTOR activator IGF-1. These results
provide a novel MGMT-independent mechanism for TMZ resistance and
highlight the clinical use of Liq to optimize TMZ therapeutics.
PI3K/AKT/mTOR signaling plays a central role in
regulating protein synthesis, proliferation and survival, and has
been implicated in multiple drug resistance in numerous cancer
types. Several components of this pathway, including epidermal
growth factor receptor, Ras, PI3K and AKT, have been observed to be
frequently mutated in tumor cells and are associated with the
hyperactivation of the pathway (31). Independent of the MGMT function,
PI3K/AKT/mTOR signaling may facilitate the expression of a set of
anti-apoptotic factors and activation of survival signals, which
rid the cells of the cytotoxic effects induced by TMZ treatment
(31,32). For example, enhanced PI3K/AKT/mTOR
activity is linked to the overexpression of c-myc, vascular
endothelial growth factor and hypoxia-inducible factor-1α (24,31).
A previous study also revealed a mechanistic link between
PI3K/AKT/mTOR hyperactivation and deregulation of homeobox A9/A10,
which underlies a drug-resistant, progenitor cell phenotype in
MGMT-independent pediatric glioblastoma (33). Moreover, there is also a
possibility that PI3K/AKT/mTOR signaling controls cell cycle
progression and mediates DNA damage repair through the
non-homologous end-joining repair pathway. A previous study
suggested that AKT interacts with the DNA-protein kinase catalytic
subunit and induces DNA double-strand break repair (34). This provides an alternative
strategy to repair TMZ-induced DNA damage and confer cell
resistance to TMZ treatment. In line with these findings, it has
been observed that the activated PI3K/AKT pathway promotes
resistance to anti-estrogen drugs in breast cancer. Loss of
phosphatase and tensin homolog (PTEN) and overexpression of PI3K
are linked to cisplatin resistance in ovarian cancer cells
(35). Previous studies also
suggest that overexpression of PIK3CA and activation of
PI3K/AKT/mTOR signaling confer trastuzumab resistance in breast
cancer (36), while constitutively
active AKT is causally linked to drug resistance against tumor
necrosis factor-related apoptosis-inducing ligand (37).
Accordingly, the PI3K/AKT/mTOR pathway is an
extremely appealing therapeutic target given its significant roles
in regulating multiple survival signaling and drug resistance
pathways. Treatment with PI3K/AKT/mTOR inhibitors may result in
growth arrest and induced cell death, while the combination of
these inhibitors with other therapeutic agents often produces
synergistic effects in the inhibition of tumor proliferation. For
example, combination treatment of TMZ with the dual PI3K/mTOR
inhibitor PI-103 resulted in a highly synergistic inhibition of
cell survival in the TMZ-resistant cell line KNS42 (33), while combined treatment of dual
PI3K/mTOR inhibitor XL765 enhanced TMZ-induced cytotoxicity in
pituitary adenoma cells and in a mouse model (29). Our results also support the
efficacy of targeting PI3K/AKT/mTOR using Liq as a strategy for
overcoming TMZ resistance in U138 cells. We suggest that TMZ
treatment in conjunction with Liq is a feasible therapy option with
several benefits. Firstly, Liq was observed to be well tolerated
with less neuronal toxicity in phase II and III clinical trials.
Secondly, Liq possesses strong blood-brain barrier permeability and
is able to reach glioma cells. Thirdly, ERβ agonist selectively
targets cells expressing ERβ, which have better specificity than
other general PI3K/mTOR inhibitors. Finally, we suggest that in
addition to its role in enhancing TMZ resistance, Liq may increase
the expression of ERβ and activate the function of this well-known
tumor suppressor to inhibit tumor proliferation. We thus suggest
that ERβ agonists are suitable for optimizing TMZ therapies and for
preventing recurrent glioma formation.
In the current study, we provide evidence that,
through ERβ knockdown or PI3K/AKT/mTOR activation using IGF-1, Liq
function on TMZ sensitivity is ERβ-dependent and
PI3K/AKT/mTOR-dependent. However, the manner in which Liq-induced
ERβ activation inhibits the activity of PI3K/AKT/mTOR signaling
requires further investigation. It has been suggested that
overexpression of ERβ is linked to upregulation of PTEN and
downregulation of human epidermal growth factor 2 (HER2)/HER3
(28), supporting a
transcription-dependent mechanism of ERβ function. Further
high-throughput studies, such as RNA-seq and proteomic studies,
will help to identify downstream targets of ERβ that are involved
in regulating the PI3K/AKT/mTOR pathway. Notably, given that PTEN
and components of the PI3K/AKT/mTOR pathway are frequently mutated
in cancer cells (38), it is
conceivable that certain cells may have a weak response to ERβ
signaling due to their lack of functionally intact proteins.
Further attempts should be made to correlate the PTEN status and
PI3K/AKT/mTOR activity with the response to the ERβ agonist in a
large number of cell types and patient samples. To conclude, the
results of the present study suggest that the ERβ agonist is a
promising therapy for overcoming TMZ chemoresistance in human
malignant glioma cells.
Acknowledgements
This study was supported in part by the National
Natural Science Foundation of China (230406).
References
|
1
|
Lacroix M, Abi-Said D, Fourney DR, et al:
A multivariate analysis of 416 patients with glioblastoma
multiforme: prognosis, extent of resection, and survival. J
Neurosurg. 95:190–198. 2001. View Article : Google Scholar
|
|
2
|
Kanu OO, Hughes B, Di C, et al:
Glioblastoma multiforme oncogenomics and signaling pathways. Clin
Med Oncol. 3:39–52. 2009.PubMed/NCBI
|
|
3
|
Stupp R, Mason WP, van den Bent MJ, et al:
Radiotherapy plus concomitant and adjuvant temozolomide for
glioblastoma. N Engl J Med. 352:987–996. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Minniti G, Salvati M, Arcella A, et al:
Correlation between O6-methylguanine-DNA methyltransferase and
survival in elderly patients with glioblastoma treated with
radiotherapy plus concomitant and adjuvant temozolomide. J
Neurooncol. 102:311–316. 2011. View Article : Google Scholar
|
|
5
|
Watanabe R, Nakasu Y, Tashiro H, et al:
O6-methylguanine DNA methyltransferase expression in tumor cells
predicts outcome of radiotherapy plus concomitant and adjuvant
temozolomide therapy in patients with primary glioblastoma. Brain
Tumor Pathol. 28:127–135. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yin S, Yang J, Lin B, et al: Exome
sequencing identifies frequent mutation of MLL2 in non-small cell
lung carcinoma from Chinese patients. Sci Rep. 4:60362014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Leung YK, Mak P, Hassan S and Ho SM:
Estrogen receptor (ER)-beta isoforms: a key to understanding
ER-beta signaling. Proc Natl Acad Sci USA. 103:13162–13167. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Burns KA and Korach KS: Estrogen receptors
and human disease: an update. Arch Toxicol. 86:1491–1504. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li LC, Yeh CC, Nojima D and Dahiya R:
Cloning and characterization of human estrogen receptor beta
promoter. Biochem Biophys Res Commun. 275:682–689. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Roepke TA, Ronnekleiv OK and Kelly MJ:
Physiological consequences of membrane-initiated estrogen signaling
in the brain. Front Biosci (Landmark Ed). 16:1560–1573. 2011.
View Article : Google Scholar :
|
|
11
|
Liu MM, Albanese C, Anderson CM, et al:
Opposing action of estrogen receptors alpha and beta on cyclin D1
gene expression. J Biol Chem. 277:24353–24360. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bailey ST, Shin H, Westerling T, Liu XS
and Brown M: Estrogen receptor prevents p53-dependent apoptosis in
breast cancer. Proc Natl Acad Sci USA. 109:18060–18065. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nilsson S and Gustafsson JA: Estrogen
receptors: therapies targeted to receptor subtypes. Clin Pharmacol
Ther. 89:44–55. 2011. View Article : Google Scholar
|
|
14
|
Paruthiyil S, Cvoro A, Zhao X, et al: Drug
and cell type-specific regulation of genes with different classes
of estrogen receptor beta-selective agonists. PLoS One.
4:e62712009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Suzuki F, Akahira J, Miura I, et al: Loss
of estrogen receptor beta isoform expression and its correlation
with aberrant DNA methylation of the 5′-untranslated region in
human epithelial ovarian carcinoma. Cancer Sci. 99:2365–2372. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mersereau JE, Levy N, Staub RE, et al:
Liquiritigenin is a plant-derived highly selective estrogen
receptor beta agonist. Mol Cell Endocrinol. 283:49–57. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kim YW, Zhao RJ, Park SJ, et al:
Anti-inflammatory effects of liquiritigenin as a consequence of the
inhibition of NF-kappaB-dependent iNOS and proinflammatory
cytokines production. Br J Pharmacol. 154:165–173. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang EJ, Park GH and Song KS:
Neuroprotective effects of liquiritigenin isolated from licorice
roots on glutamate-induced apoptosis in hippocampal neuronal cells.
Neurotoxicology. 39:114–123. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhou M, Higo H and Cai Y: Inhibition of
hepatoma 22 tumor by Liquiritigenin. Phytother Res. 24:827–833.
2010.
|
|
20
|
Sareddy GR, Nair BC, Gonugunta VK, et al:
Therapeutic significance of estrogen receptor beta agonists in
gliomas. Mol Cancer Ther. 11:1174–1182. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ryu CH, Yoon WS, Park KY, et al: Valproic
acid downregulates the expression of MGMT and sensitizes
temozolomide-resistant glioma cells. J Biomed Biotechnol.
2012:9874952012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Vladusic EA, Hornby AE, Guerra-Vladusic
FK, Lakins J and Lupu R: Expression and regulation of estrogen
receptor beta in human breast tumors and cell lines. Oncol Rep.
7:157–167. 2000.
|
|
23
|
Hennessy BT, Smith DL, Ram PT, Lu Y and
Mills GB: Exploiting the PI3K/AKT pathway for cancer drug
discovery. Nat Rev Drug Discov. 4:988–1004. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shaw RJ and Cantley LC: Ras, PI(3)K and
mTOR signalling controls tumour cell growth. Nature. 441:424–430.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yin S, Wang P, Deng W, et al: Dosage
compensation on the active X chromosome minimizes transcriptional
noise of X-linked genes in mammals. Genome Biol. 10:R742009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yin S, Deng W, Hu L and Kong X: The impact
of nucleosome positioning on the organization of replication
origins in eukaryotes. Biochem Biophys Res Commun. 385:363–368.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li W, Winters A, Poteet E, et al:
Involvement of estrogen receptor beta5 in the progression of
glioma. Brain Res. 1503:97–107. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lindberg K, Helguero LA, Omoto Y,
Gustafsson JA and Haldosen LA: Estrogen receptor beta represses Akt
signaling in breast cancer cells via downregulation of HER2/HER3
and upregulation of PTEN: implications for tamoxifen sensitivity.
Breast Cancer Res. 13:R432011. View
Article : Google Scholar
|
|
29
|
Dai C, Zhang B, Liu X, et al: Inhibition
of PI3K/AKT/mTOR pathway enhances temozolomide-induced cytotoxicity
in pituitary adenoma cell lines in vitro and xenografted pituitary
adenoma in female nude mice. Endocrinology. 154:1247–1259. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Myers MG Jr, Grammer TC, Wang LM, et al:
Insulin receptor substrate-1 mediates phosphatidylinositol
3′-kinase and p70S6k signaling during insulin, insulin-like growth
factor-1, and interleukin-4 stimulation. J Biol Chem.
269:28783–28789. 1994.PubMed/NCBI
|
|
31
|
Jiang BH and Liu LZ: Role of mTOR in
anticancer drug resistance: perspectives for improved drug
treatment. Drug Resist Updat. 11:63–76. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Deng WJ, Nie S, Dai J, Wu JR and Zeng R:
Proteome, phosphoproteome, and hydroxyproteome of liver
mitochondria in diabetic rats at early pathogenic stages. Mol Cell
Proteomics. 9:100–116. 2010. View Article : Google Scholar :
|
|
33
|
Gaspar N, Marshall L, Perryman L, et al:
MGMT-independent temozolomide resistance in pediatric glioblastoma
cells associated with a PI3-kinase-mediated HOX/stem cell gene
signature. Cancer Res. 70:9243–9252. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Toulany M, Lee KJ, Fattah KR, et al: Akt
promotes post-irradiation survival of human tumor cells through
initiation, progression, and termination of DNA-PKcs-dependent DNA
double-strand break repair. Mol Cancer Res. 10:945–957. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chock KL, Allison JM, Shimizu Y and
ElShamy WM: BRCA1-IRIS overexpression promotes cisplatin resistance
in ovarian cancer cells. Cancer Res. 70:8782–8791. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
O’Brien NA, Browne BC, Chow L, et al:
Activated phosphoinositide 3-kinase/AKT signaling confers
resistance to trastuzumab but not lapatinib. Mol Cancer Ther.
9:1489–1502. 2010. View Article : Google Scholar
|
|
37
|
Xu J, Zhou JY, Wei WZ and Wu GS:
Activation of the Akt survival pathway contributes to TRAIL
resistance in cancer cells. PLoS One. 5:e102262010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang SI, Puc J, Li J, et al: Somatic
mutations of PTEN in glioblastoma multiforme. Cancer Res.
57:4183–4186. 1997.PubMed/NCBI
|