Introduction
Overproduction of reactive oxygen species (ROS),
such as hydrogen peroxide (H2O2), may induce
pathological hepatocyte apoptosis (1). Oxidant-induced apoptosis has a
pivotal role in the development and progression of liver diseases,
including alcoholic liver diseases (2), drug-induced liver injury (3), viral hepatitis (4,5),
cholestatic liver diseases (6),
non-alcoholic steatohepatitis (7)
and ischemic/reperfusion injury, and also contributes to liver
fibrogenesis (8). Therefore, the
inhibition of hepatocyte apoptosis may be a promising novel
therapeutic choice for the treatment of liver injury and
fibrosis.
Apoptosis is initiated through two fundamental
pathways: The extrinsic pathway, mediated by death receptors, and
the intrinsic pathway, initiated by mitochondrial dysfunction
(9). The cysteine aspartate
protease (caspase) family of enzymes are key molecules whose
activation may result in apoptosis; these include upstream
initiator caspases (e.g. caspase-8 and -9) and downstream effector
caspases (e.g. caspase-3). In addition, ROS-induced endoplasmic
reticulum stress (ERS) may initiate pathways which lead to caspase
activation and apoptosis (10).
The primary ERS-associated pathways are activated by endoplasmic
reticulum (ER) membrane-associated proteins, including protein
kinase R-like ER kinase (PERK), inositol-requiring enzyme 1 and
activating transcription factor 6. Each of these pathways
upregulates the transcription factor CCAAT-enhancer-binding protein
homologous protein (CHOP), which results in decreased expression of
anti-apoptotic B cell lymphoma (Bcl)-2 (11) and increased expression of
pro-apoptotic Bcl-2 interacting mediator of cell death (12), therefore inducing apoptosis. The
PERK pathway also activates caspase-12, which directly cleaves
procaspase-9 and then activates caspase-3, resulting in apoptosis
(13).
Relaxin-3, first identified in 2002, is the
ancestral peptide of the human relaxin subclass of the insulin
superfamily (14). The primary
site of relaxin-3 messenger (m)RNA expression is the brain;
however, relaxin-3 is also present in other tissues, such as the
liver (14). At present, the roles
and mechanism of action of relaxin-3 remain to be fully elucidated;
one study suggested that relaxin-3 was involved in brain functions,
including the stress response and regulation of food intake, in
combination with relaxin family peptide receptor 3 [RXFP-3/G
protein-couple receptor (GPCR) 135] (15). In addition, relaxin-3 was also
reported to bind to RXFP-1/leucine-rich repeat-containing GPCR 7
(LGR7; the primary receptor of relaxin-2) (16). In a previous study, LGR7 was
expressed at a low level in normal rat livers; however, cirrhotic
rat livers expressed significantly increased LGR7 levels in active
hepatic stellate cells (17).
Relaxin-3 treatment was reported to significantly increase the
production of cyclic adenosine monophosphate, indicating the role
of relaxin-3 in liver injury protection (18). Previous studies demonstrated that
relaxin-2, in combination with LGR7, inhibited apoptosis in
reproductive organ tissues during pregnancy (19) and in the heart (20). It was therefore hypothesized that
relaxin-3 may attenuate hepatocyte apoptosis and protect against
liver injury. The present study aimed to investigate the direct
effect of relaxin-3 on hepatocyte apoptosis and its mechanism of
action.
Materials and methods
Reagents
Synthetic human relaxin-3 was obtained from Phoenix
Pharmaceuticals, Inc. (Burlingame, CA, USA). Rabbit anti-human CHOP
polyclonal antibody as well as rabbit anti-human cleaved caspase 8
and 12 polyclonal antibodies were purchased from Abcam (Cambridge,
UK). Rabbit anti-human Beclin 1 polyclonal antibody as well as
rabbit anti-human cleaved caspase 9 and 3 antibodies were purchased
from Cell Signaling Technologies, Inc. (Beverly, MA, USA). Rabbit
anti-human microtubule associated protein 1 light chain 3 (LC3)
polyclonal antibodies were purchased from Sigma (St. Louis, MO,
USA). All chemicals and reagents used in this study were of
analytical grade. The human normal liver cell line L02 was
purchased from the cell bank of the Institute of Biochemistry and
Cell Biology (Shanghai, China).
Hepatocyte culture and treatment
Human hepatocyte L02 cells were cultured at 37°C in
an incubator (5% CO2, 95% air). RPMI-1640 medium
(Hyclone, Inc., Logan, UT, USA) supplemented with 10% fetal bovine
serum (Sijiqing, Inc., Huzhou, China) was used for the cell
cultures. A dose-response experiment was initially conducted on the
cells, which were treated with 0, 20, 50, 100, 200, 400, 600, 800
and 1,000 μmol/l H2O2 (Sigma); 200 μmol/l was
selected as the optimal dose for all subsequent experiments. At
~70–80% confluence, cells were divided into the following five
groups: Control; H2O2, 200 μmol/l
H2O2; R10, 200 μmol/l
H2O2 + 10 ng/ml relaxin-3; R50, 200 μmol/l
H2O2 + 50 ng/ml relaxin-3; and R100, 200
μmol/l H2O2 + 100 ng/ml relaxin-3. Following
co-incubation for 24 h, cells from all five groups were collected
for analysis.
Cell viability
Relative cell viability was determined using an MTT
assay (Sigma). Cells were plated in 96-well microtiter plates
(Corning, Inc., New York, NY, USA). Following cell treatment, MTT
was added to the culture medium to yield a final MTT concentration
of 0.5 mg/ml; cells were then incubated for 4 h at 37°C. The dye
was dissolved by adding dimethyl sulfoxide at room temperature for
10 min. The preparations were agitated thoroughly with the cells
containing formazan crystals using a plate shaker. Absorbance was
measured at 490 nm using a microplate reader (Bio-Rad, Inc.,
Hercules, CA, USA).
Hoechst staining
Apoptotic cells were characterized due to a
distinctive condensed nuclear structure following staining with
Hoechst 33258 (Beyotime, Inc., Haimen, China) and visible
chromosomal fragmentation. Treated cells were fixed with 4%
paraformaldehyde (Westang, Inc., Shanghai, China) for 15 min at
room temperature, washed in phosphate-buffered saline (PBS) and
then stained with Hoechst dye for 20 min at room temperature in the
dark. Following washing with PBS, blue fluorescent cells were
examined under a confocal scanning laser microscope (Nikon, Inc.,
Tokyo, Japan).
Transmission electron microscopy
Cells were harvested and fixed using 3.0%
glutaraldehyde and 1.5% paraldehyde. Cells were then washed in PBS,
fixed in osmium tetroxide (Xiya, Inc., Chengdu, China) and then
dehydrated in an ethanol series. Subsequently, the samples were
embedded in epoxy resin and examined under a transmission electron
microscope (Olympus, Inc., Tokyo, Japan).
Western blot analysis
Following treatment for 24 h, cells were washed with
PBS and resuspended in cold lysis buffer containing
phenylmethylsulfonyl fluoride. Cell lysates were incubated on ice
for 30 min and then centrifuged at 12,000 xg for 15 min at 4°C. The
protein content of the supernatant was determined using a
bicinchoninic acid-200 protein assay kit (Beyotime, Inc.). Equal
amounts of protein (20 μg) from each group were separated using 12%
SDS-PAGE (Sanland, Inc., Xiamen, China) and transferred onto
polyvinylidene difluoride (PVDF) membranes (Gelman, Inc., Morgan
Hill, CA, USA). The membranes were blocked using 5% skimmed milk
(Yili, Inc., Neimenggu, China) for 1 h at room temperature with
agitation and then incubated with the corresponding primary
antibodies overnight at 4°C. All of the following primary
antibodies were diluted in Tris-buffered saline/Tween 20 (TBST)
solution (anti-β-actin, 1:2,000; anti-CHOP, 1:500; anti-cleaved
caspase-12, 1:500; anti-cleaved caspase-8, 1:1,000; anti-cleaved
caspase-9, 1:1,000; anti-cleaved caspase-3, 1:1,000; anti-Beclin-1,
1:1,000; and anti-LC3, 1:1,000). Samples were then washed three
times (10 min/wash) with TBST and a secondary antibody (1:2,000)
conjugated with alkaline phosphatase (ZSGB-BIO, Inc., Beijing,
China) was added to the membranes, which were then incubated at
room temperature for 1 h with agitation. The membranes were then
washed three times (10 min/wash) prior to visualization using
Western Blue® stabilized substrate for alkaline
phosphatase (Promega Corp., Madison, WI, USA). In order to quantify
the band intensities, the western blot membranes were scanned using
ImageJ software (National Institute of Health, Bethesda, MA, USA).
The results were normalized based on the respective levels of
β-actin.
Statistical analysis
GraphPad Prism software version 5.0 (La Jolla, CA,
USA) was used for data analysis. Each experiment was repeated a
minimum of three times and values are expressed as the mean ±
standard deviation. The one-way analysis of variance followed by
the Newman-Keuls multiple comparison test were used for comparisons
among the five groups. P<0.05 was considered to indicate a
statistically significant difference between values.
Results
Relaxin-3 attenuates
H2O2-induced hepatocyte apoptosis
Cell viability assays, Hoechst staining, electron
microscopy and western blot analysis were used in order to
determine the effect of treatment with graded concentrations of
relaxin-3 on H2O2-induced hepatocyte
apoptosis.
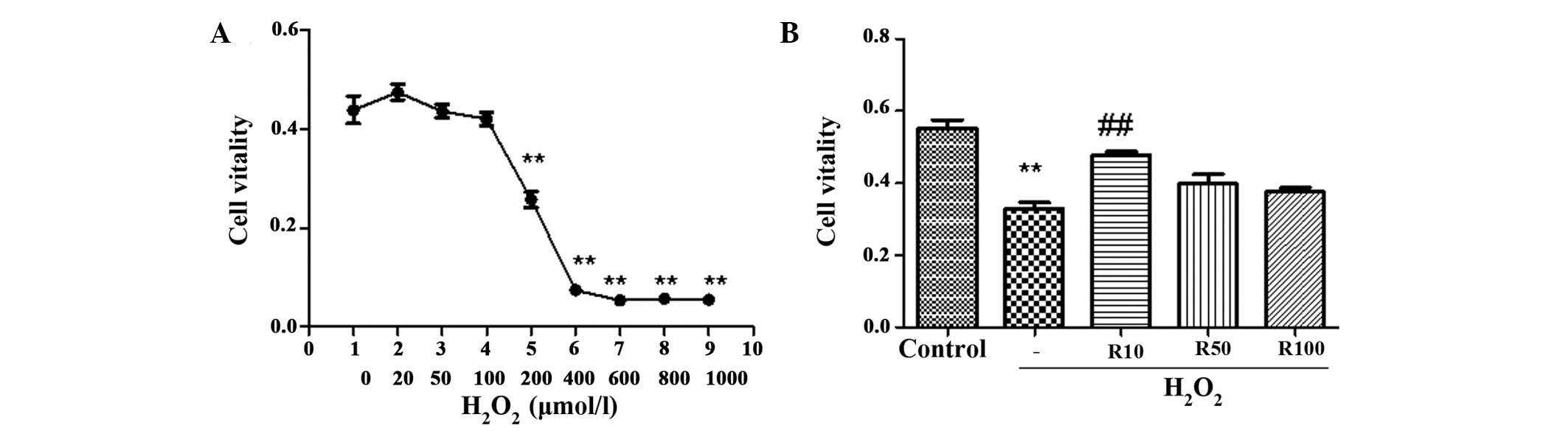
As shown in Fig.
1A, exposure of human hepatocytes to graded concentrations of
H2O2 (0–1,000 μmol/l) for 24 h remarkably
decreased cell viability in a dose-dependent manner. Concentrations
of H2O2 produced significant results at 200
μmol/l and reached maximum inhibition between 400–600 μmol.
Administration of 10 ng/ml relaxin-3 to cells treated with 200 μmol
H2O2 was found to significantly enhance cell
viability; however, the protective effect of relaxin-3 diminished
at higher doses (Fig. 1B).
Nuclear condensation, a hallmark characteristic of
apoptotic cells, was observed in cells in the
H2O2 only group following Hoechst staining.
By contrast, cells treated with relaxin-3 were round and
homogeneous, with a markedly decreased number of apoptotic nuclei,
most notably in the R10 group (Fig.
2A). Cell morphological changes were also observed using
electron microscopy (Fig. 2B). The
later stages of apoptosis are characterized by nuclear pyknosis,
mitochondrial and endoplasmic reticulum distension, as well as the
formation of apoptotic bodies. These characteristics of later stage
apoptosis were observed in cells in the H2O2
group; however, no obvious apoptotic changes were detected in the
relaxin-3 treated cells. Quantification of the percentage of
apoptotic cells counted in each group by Hoechst staining is shown
in Fig. 2C. The results
demonstrated that H2O2 treatment
significantly increased the number of apoptotic cells compared with
that of the control. By contrast, the R10 relaxin-3 treatment group
(10 ng/ml) showed a significantly decreased number of apoptotic
cells compared with that of the H2O2 group
(P<0.05); however, the decrease in the number of apoptotic cells
in the R50 and R100 treatment groups were not significant compared
with levels in the H2O2 group (Fig. 2C).
 | Figure 2Relaxin-3 inhibits
H2O2-induced hepatocyte apoptosis. (A)
Hoechst staining reveals condensed and fragmented apoptotic nuclei
of hepatocytes in the control, H2O2 or
relaxin-3-treated (R10, R50 and R100 for 24 h) groups. Arrows
indicate condensed nuclei (magnification, ×100; n=5). (B)
Transmission electron microscopy of hepatocyte apoptosis in each
group (magnification, ×10,000). (C) Quantification of the
percentage of apoptotic cells counted in each group. (D) Western
blot analysis and quantification of cleaved caspase-3 protein
levels in each group. β-actin was used as the internal control.
Values are presented as the mean ± standard deviation (n=3).
*P<0.05 vs. control, **P<0.01 vs.
control, #P<0.05 vs. H2O2,
##P<0.01 vs. H2O2.
H2O2, hydrogen peroxide treatment;
R10/50/100, treatment with 10, 50 or 100 ng/ml relaxin-3,
respectively. |
Intrinsic and extrinsic apoptotic pathways activate
the proenzyme form of caspase-3, which is then cleaved through
self-proteolysis and other proteases (21). Therefore, endogenous cleaved
(activated) caspase-3 is a common indicator of cellular apoptosis.
In the present study, western blot analysis was used to determine
the expression levels of cleaved caspase-3. The results
demonstrated that H2O2 treatment
significantly increased the expression of cleaved caspase-3
compared to that of the control. By contrast, the relaxin-3 R10
treatment group (10 ng/ml) showed significantly attenuated cleaved
caspase-3 protein levels compared to those of the
H2O2 group (P<0.05); however, the decrease
in caspase-3 protein levels in the R50 and R100 treatment groups
was not significant compared to levels in the
H2O2 group (Fig.
2D).
Relaxin-3 attenuates the
H2O2-induced increase in cleaved caspase-8
and -9 levels
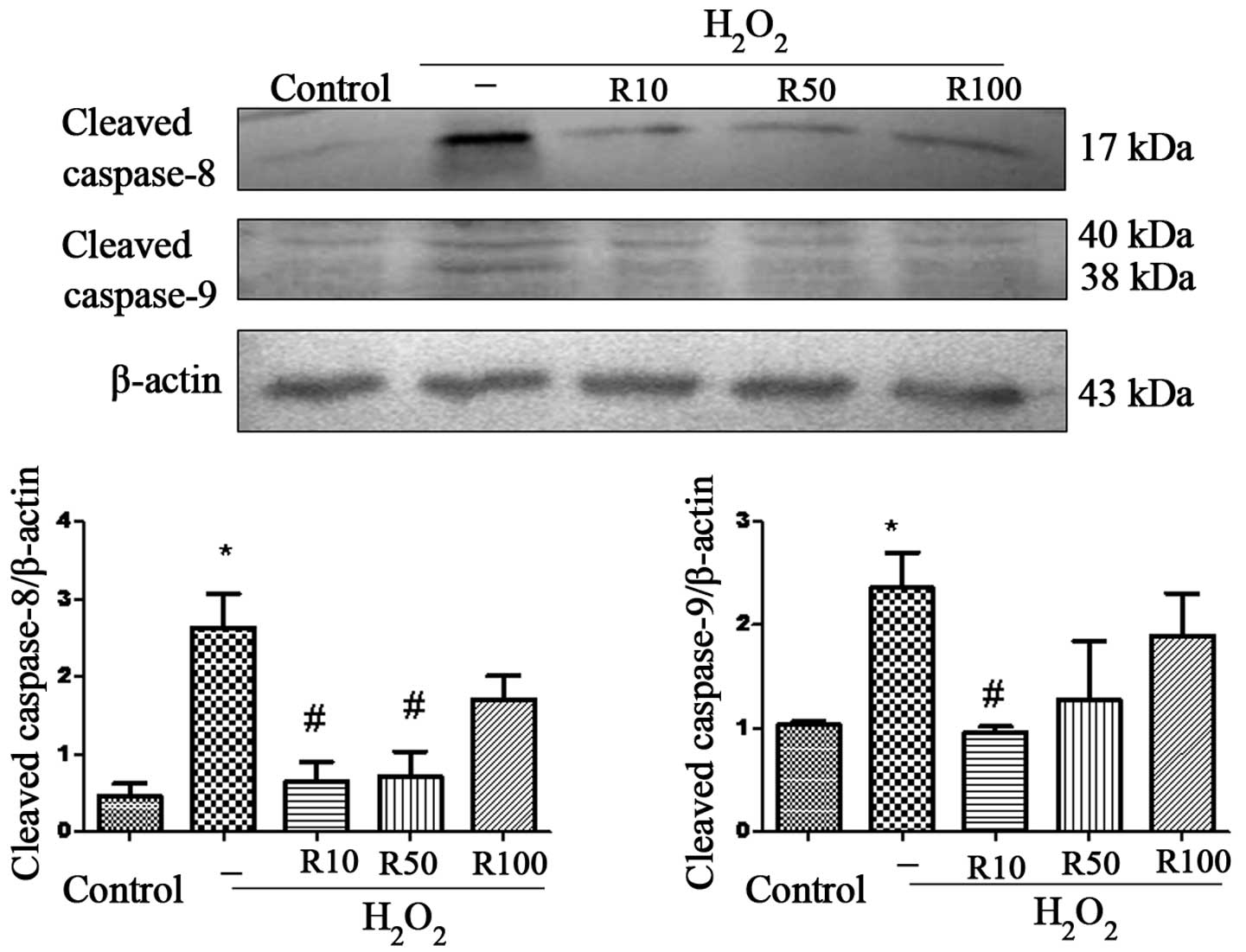
Caspase-8 and -9 are upstream mediators of the
extrinsic and intrinsic apoptotic pathways, respectively; cleavage
of these caspases results in the activation of downstream
executioner caspases, such as caspase-3 (21). Therefore, in the present study,
western blot analysis was used to determine the levels of cleaved
caspase-8 and -9 using western blot anlaysis and investigate
whether these pathways were involved in the anti-apoptotic effect
of relaxin-3 (Fig. 3). The results
revealed that the protein expression of cleaved caspase-8 and -9
was significantly increased in the H2O2 group
compared to that of the control group. However, the R10 and R50
relaxin-3 treatment groups demonstrated significantly attenuated
cleaved caspase-8 levels compared to those of the
H2O2 group; in addition, cleaved caspase-9
protein expression was significantly reduced in the R10 group
(P<0.05) .
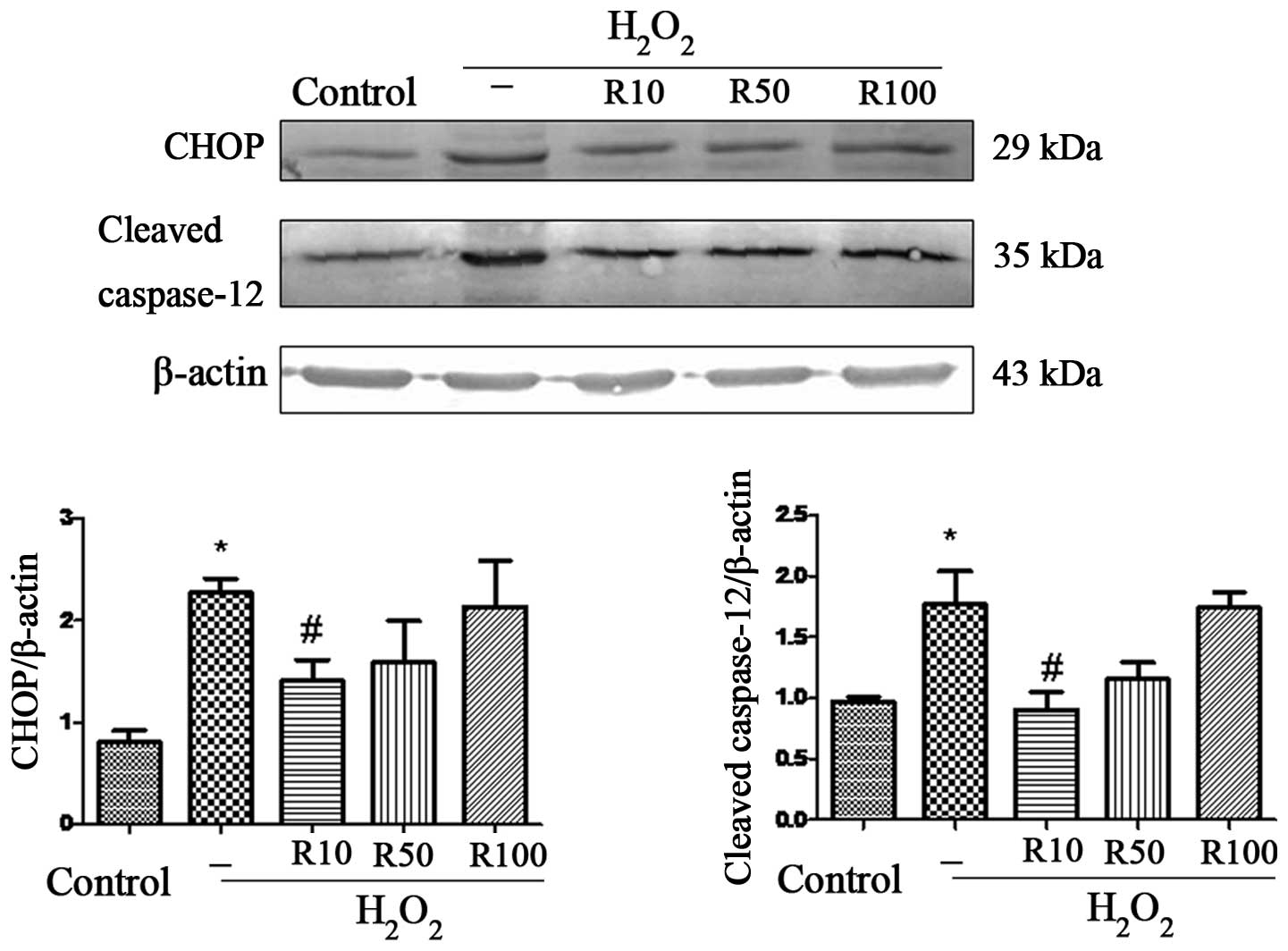
Relaxin-3 inhibits
H2O2-induced ERS
Sustained or severe ERS leads to apoptosis through
several pathways, including the CHOP and caspase-12 pathways
(22). Therefore, the present
study used western blot analysis to detect protein expression
levels of CHOP and cleaved caspase-12 in order to directly examine
the role of relaxin-3 in ERS-associated apoptosis (Fig. 4). The results revealed elevated
levels of CHOP and cleaved caspase-12 in the
H2O2 group. However, 10 μg/ml relaxin-3
treatment significantly inhibited the
H2O2-induced overexpression of CHOP and
cleaved caspase-12 (P<0.05), while this effect was not observed
at higher doses of relaxin-3.
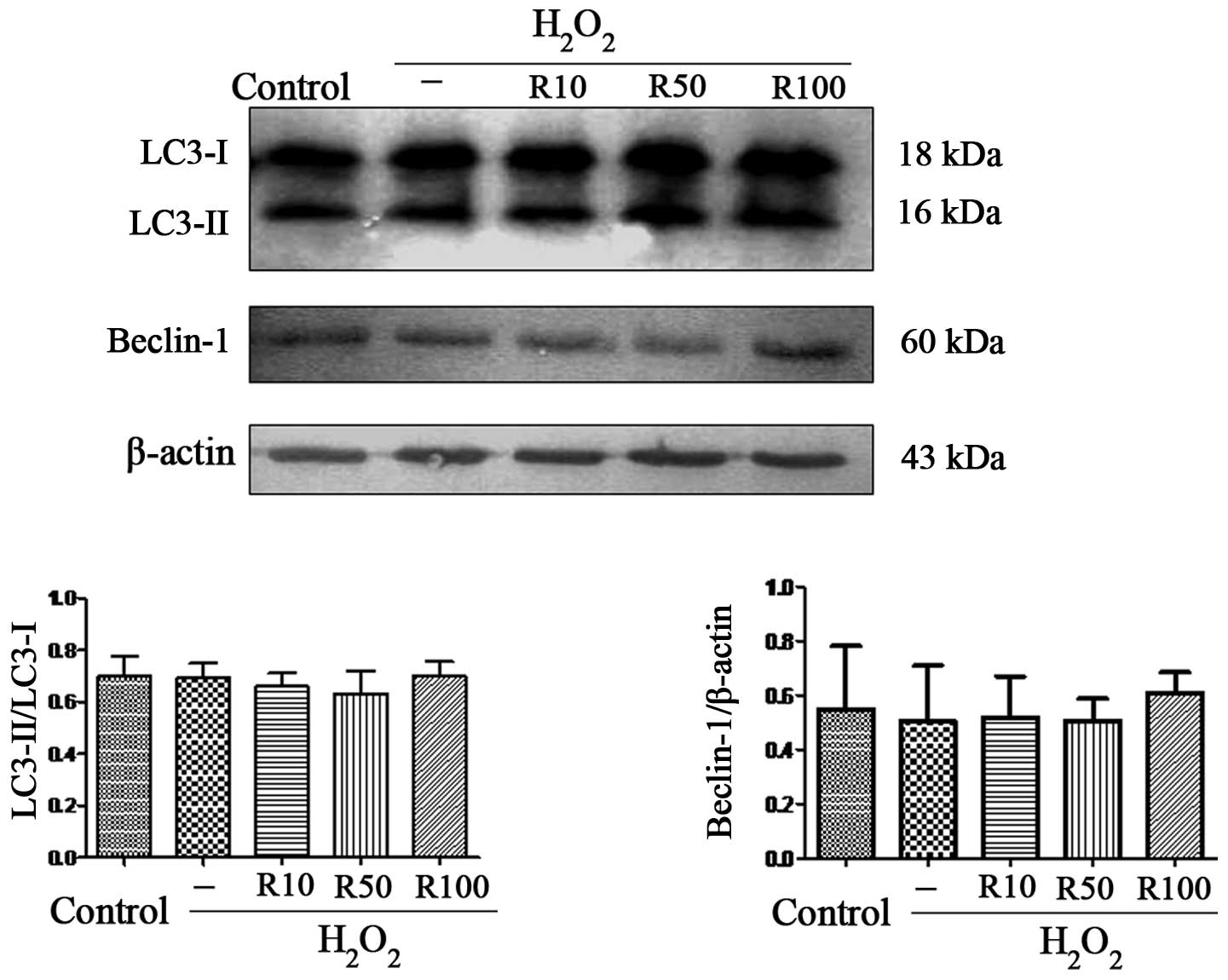
Relaxin-3 has no effect on markers of
autophagy in apoptotic hepatocytes
Autophagy is the process by which cells recycle
cytoplasm and degrade surplus or dysfunctional organelles (23). It has been reported that autophagy
was the cell survival mechanism of primary human hepatocytes during
oxidative stress (24). In order
to examine the effect of relaxin-3 on autophagy in
H2O2-induced hepatocyte apoptosis, the
present study aimed to determine the expression levels of
LC3-II/LC3-I and Beclin-1. The results showed that the expression
of these autophagy markers was unchanged following
H2O2 treatment alone or in combination with
relaxin-3 treatment (Fig. 5).
Discussion
Relaxin-3, the ‘ancestral’ member of the relaxin
peptide family, activates RXFP3 which is involved in stress and
anxiety responses (25) as well as
appetite regulation (26,27), and whose expression is highest in
the brain (28). However, the
effect of relaxin-3 on other organs and tissues is has not been
studied in detail (29). To the
best of our knowledge, the present study was the first to
demonstrate the impact of relaxin-3 on
H2O2-induced hepatocyte apoptosis.
H2O2 is widely used as a model
of oxidative stress in order to induce apoptosis.
H2O2 stimulation induces enhances expression
of ROS, which may exhaust hepatocellular antioxidant defenses,
resulting in oxidative stress and hepatocyte apoptosis (30). In the present study, hepatocytes
were incubated with 200 μmol/l H2O2 for 24 h,
as previously reported (31).
Following treatment with relaxin-3 (10 ng/ml), a significant
increase in cell viability was observed as well as a marked
decrease in cleaved caspase-3 expression compared with levels in
the untreated H2O2 group. In addition,
Hoechst staining and electron microscopy revealed a decrease in the
number of apoptotic hepatocytes. The results therefore indicated
that relaxin-3 was involved in the protection of hepatocytes from
pathological apoptosis.
The role of relaxin-3 in stress/anxiety and appetite
regulation is currently defined by the regional distribution of
RXFP3 in the brain, and relaxin-3 was also reported to bind to
RXFP1, a relaxin-2 receptor, which is expressed primarily in the
liver (16,32). A previous study reported that
relaxin-3 had an anti-fibrotic effect on cirrhotic liver tissue,
which resulted in RXFR1 upregulation (18). In addition, the anti-apoptotic
action of relaxin-2 in combination with RXFP1 was previously
described in cardiac (20) and
reproductive tissues (19,33). Based on evidence provided by these
previous studies, it was hypothesized that relaxin-3 may attenuate
hepatocyte apoptosis via the activation of RXFP1; however, further
studies are required in order to characterize the role of RXFPs in
apoptotic hepatocytes and determine the signaling properties of
relaxin-3.
Of note, in the present study, the anti-apoptotic
action of relaxin-3 did not proceed in a dose-dependent manner; by
contrast, the low dose of relaxin-3 (10 ng/ml) produced significant
results, indicative of its effectiveness in protecting hepatocytes
from apoptosis, while higher concentrations of relaxin-3 (50 and
100 ng/ml) were less effective and produced non-significant results
in the majority of experiments performed. Further studies are
required in order to elucidate the mechanisms underlying the
effectiveness of low-dose relaxin-3 compared to that of higher
doses.
Numerous studies have confirmed that caspases have a
pivotal role in H2O2-induced apoptosis. There
are two major pathways by which hepatocyte apoptosis is induced
(34): The extrinsic (death
receptor-mediated) pathway, which is initiated through the
activation of caspase-8 by death receptors followed by the
caspase-8 induced activation of an effector caspase (caspase-3);
the intrinsic (mitochondrial apoptosis) pathway, which is initiated
by the activation of caspase-9 following cytochrome c release from
mitochondria, which in turn activates caspase-3. Previous studies
have associated H2O2-mediated apoptosis with
the intrinsic pathway of hepatocyte apoptosis (30,35);
however, the results of the present study demonstrated the
significantly increased expression of cleaved caspase-8 as well as
caspase-9 following hepatocyte treatment with
H2O2. These results may be explained by the
close interlinking of the two pathways, as the mitochondrial
pathway may amplify the relatively weak death receptor-induced
apoptotic signal in hepatocytes (36). Furthermore, relaxin-3 treatment was
found to significantly decrease the expression of cleaved caspase-8
and -9, indicating that relaxin-3 prevented hepatocyte apoptosis
via the inhibition of caspase-8 and -9 activation.
The ER, analogous to mitochondria, directly
initiates caspase activation and apoptotic pathways (10). A previous study reported that ERS
was involved in H2O2-induced apoptosis in
hepatocytes (37), which is
consistent with the results of the present study, as levels of
cleaved caspase-12 and CHOP were markedly increased following
incubation with H2O2. In addition, relaxin-3
significantly reduced the overexpression of cleaved caspase-12 and
CHOP, providing evidence for the attenuation of
H2O2-induced hepatocyte apoptosis following
relaxin-3 treatment via the inhibition of ERS.
Autophagy is an important mechanism by which cells
maintain homeostasis; however, the precise role of autophagy within
hepatocytes during liver disease and injury remains to be fully
elucidated. A recent study reported that hepatocyte autophagy
served as a cell survival mechanism for primary human hepatocytes
under hypoxic stress (24).
However, the results of the present study revealed that
H2O2 and relaxin-3 did not induce any obvious
changes in the expression of hepatocyte autophagy markers. This
therefore indicated that autophagy was not involved in the late
stages of severe hepatocyte injury, which contradicts the results
of a previous study, which reported that mild ischemia led to the
induction of autophagy and apoptosis, while moderate/severe
ischemia induced apoptotic and necrotic cell death without
increasing autophagy (38). The
morphological observations of the present study revealed that the
H2O2-treated cells showed charateristics of
the late stages of apoptosis; therefore suggesting that autophagy
may not be the major mechanism mediating late-stage cell death in
hepatocytes.
In conclusion, to the best of our knowledge, the
present study provided the first evidence for the protective effect
of relaxin-3 in hepatocytes following
H2O2-induced apoptosis. Furthermore, the
anti-apoptotic role of relaxin-3 was demonstrated to be mediated
via decreased activation of upstream initiator caspases (caspase-8
and -9) and the inhibition of the ERS pathway.
Acknowledgements
The authors would like to thank Professors Donghui
Li and Lovedip S. Kooner for their assistance in revising the
manuscript.
References
|
1
|
Horvathova E, Eckl PM, Bresgen N and
Slamenova D: Evaluation of genotoxic and cytotoxic effects of H2O2
and DMNQ on freshly isolated rat hepatocytes; protective effects of
carboxymethyl chitin-glucan. Neuro Endocrinol Lett. 29:644–648.
2008.PubMed/NCBI
|
|
2
|
Miñana JB, Gómez-Cambronero L, Lloret A,
et al: Mitochondrial oxidative stress and CD95 ligand: a dual
mechanism for hepatocyte apoptosis in chronic alcoholism.
Hepatology. 35:1205–1214. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jaeschke H, McGill MR and Ramachandran A:
Oxidant stress, mitochondria, and cell death mechanisms in
drug-induced liver injury: lessons learned from acetaminophen
hepatotoxicity. Drug Metab Rev. 44:88–106. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kundu D, Roy A, Mandal T, Bandyopadhyay U,
Ghosh E and Ray D: Oxidative stress in alcoholic and viral
hepatitis. N Am J Med Sci. 4:412–415. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Koike K: Oxidative stress and apoptosis in
hepatitis C: the core issue. J Gastroenterol. 41:292–294. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rodrigues CM, Fan G, Wong PY, Kren BT and
Steer CJ: Ursodeoxycholic acid may inhibit deoxycholic acid-induced
apoptosis by modulating mitochondrial transmembrane potential and
reactive oxygen species production. Mol Med. 4:165–178.
1998.PubMed/NCBI
|
|
7
|
Canbay A, Gieseler RK, Gores GJ and Gerken
G: The relationship between apoptosis and non-alcoholic fatty liver
disease: an evolutionary cornerstone turned pathogenic. Z
Gastroenterol. 43:211–217. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Canbay A, Friedman S and Gores GJ:
Apoptosis: the nexus of liver injury and fibrosis. Hepatology.
39:273–278. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Matthews GM, Newbold A and Johnstone RW:
Intrinsic and extrinsic apoptotic pathway signaling as determinants
of histone deacetylase inhibitor antitumor activity. Adv Cancer
Res. 116:165–197. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Breckenridge DG, Germain M, Mathai JP,
Nguyen M and Shore GC: Regulation of apoptosis by endoplasmic
reticulum pathways. Oncogene. 22:8608–8618. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
McCullough KD, Martindale JL, Klotz LO, Aw
TY and Holbrook NJ: Gadd153 sensitizes cells to endoplasmic
reticulum stress by down-regulating Bcl2 and perturbing the
cellular redox state. Mol Cell Biol. 21:1249–1259. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Puthalakath H, O’Reilly LA, Gunn P, Lee L,
et al: ER stress triggers apoptosis by activating BH3-only protein
Bim. Cell. 129:1337–1349. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nakagawa T, Zhu H, Morishima N, Li E, Xu
J, Yankner BA and Yuan J: Caspase-12 mediates
endoplasmic-reticulum-specific apoptosis and cytotoxicity by
amyloid-beta. Nature. 403:98–103. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bathgate RA, Samuel CS, Burazin TC, et al:
Human relaxin gene 3 (H3) and the equivalent mouse relaxin (M3)
gene: Novel members of the relaxin peptide family. J Biol Chem.
277:1148–1157. 2002. View Article : Google Scholar
|
|
15
|
McGowan BM, Stanley SA, Ghatei MA and
Bloom SR: Relaxin-3 and its role in neuroendocrine function. Ann NY
Acad Sci. 1160:250–255. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chan LJ, Hossain MA, Samuel CS, Separovic
F and Wade JD: The relaxin peptide family - structure, function and
clinical applications. Protein Pept Lett. 18:220–229. 2011.
View Article : Google Scholar
|
|
17
|
Fallowfield JA, Hayden AL, Snowdon VK,
Aucott RL, Stutchfield BM, Mole DJ, Pellicoro A, Gordon-Walker TT,
Henke A, Schrader J, Trivedi PJ, Princivalle M, Forbes SJ, Collins
JE and Iredale JP: Relaxin modulates human and rat hepatic
myofibroblast function and ameliorates portal hypertension in vivo.
Hepatology. 59:1492–1504. 2014. View Article : Google Scholar
|
|
18
|
Bennett RG, Dalton SR, Mahan KJ,
Gentry-Nielsen MJ, Hamel FG and Tuma DJ: Relaxin receptors in
hepatic stellate cells and cirrhotic liver. Biochem Pharmacol.
73:1033–1040. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yao L, Agoulnik AI, Cooke PS, Meling DD
and Sherwood OD: Relaxin acts on stromal cells to promote
epithelial and stromal proliferation and inhibit apoptosis in the
mouse cervix and vagina. Endocrinology. 149:2072–2079. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Moore XL, Tan SL, Lo CY, et al: Relaxin
antagonizes hypertrophy and apoptosis in neonatal rat
cardiomyocytes. Endocrinology. 148:1582–1589. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wu CC and Bratton SB: Regulation of the
intrinsic apoptosis pathway by reactive oxygen species. Antioxid
Redox Signal. 19:546–558. 2013. View Article : Google Scholar :
|
|
22
|
Shore GC, Papa FR and Oakes SA: Signaling
cell death from the endoplasmic reticulum stress response. Curr
Opin Cell Biol. 23:143–149. 2011. View Article : Google Scholar :
|
|
23
|
Ryter SW, Cloonan SM and Choi AM:
Autophagy: a critical regulator of cellular metabolism and
homeostasis. Mol Cells. 36:7–16. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bhogal RH, Weston CJ, Curbishley SM, Adams
DH and Afford SC: Autophagy: a cyto-protective mechanism which
prevents primary human hepatocyte apoptosis during oxidative
stress. Autophagy. 8:545–558. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tanaka M, Iijima N, Miyamoto Y, Fukusumi
S, Itoh Y, Ozawa H and Ibata Y: Neurons expressing relaxin 3/INSL 7
in the nucleus incertus respond to stress. Eur J Neurosci.
21:1659–1670. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
McGowan BM, Stanley SA, Smith KL, et al:
Central relaxin-3 administration causes hyperphagia in male Wistar
rats. Endocrinology. 146:3295–3300. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hida T, Takahashi E, Shikata K, et al:
Chronic intracerebroventricular administration of relaxin-3
increases body weight in rats. J Recept Signal Transduct Res.
26:147–158. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sutton SW, Bonaventure P, Kuei C, Roland
B, Chen J, Nepomuceno D, Lovenberg TW and Liu C: Distribution of
G-protein-coupled receptor (GPCR)135 binding sites and receptor
mRNA in the rat brain suggests a role for relaxin-3 in
neuroendocrine and sensory processing. Neuroendocrinology.
80:298–307. 2004. View Article : Google Scholar
|
|
29
|
Smith CM, Ryan PJ, Hosken IT, Ma S and
Gundlach AL: Relaxin-3 systems in the brain - the first 10 years. J
Chem Neuroanat. 42:262–275. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Czaja MJ: Induction and regulation of
hepatocyte apoptosis by oxidative stress. Antioxid Redox Signal.
4:759–767. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang H, Xue Z, Wang Q, Feng X and Shen Z:
Propofol protects hepatic L02 cells from hydrogen peroxide-induced
apoptosis via activation of extracellular signal-regulated kinases
pathway. Anesth Analg. 107:534–540. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
van der Westhuizen ET, Halls ML, Samuel
CS, Bathgate RA, Unemori EN, Sutton SW and Summers RJ: Relaxin
family peptide receptors - from orphans to therapeutic targets.
Drug Discov Today. 13:640–651. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhao S, Fields PA and Sherwood OD:
Evidence that relaxin inhibits apoptosis in the cervix and the
vagina during the second half of pregnancy in the rat.
Endocrinology. 142:2221–2229. 2001.PubMed/NCBI
|
|
34
|
Ghavami S, Hashemi M, Kadkhoda K, Alavian
SM, Bay GH and Los M: Apoptosis in liver diseases - detection and
therapeutic applications. Med Sci Monit. 11:RA337–RA345.
2005.PubMed/NCBI
|
|
35
|
Kannan K and Jain SK: Oxidative stress and
apoptosis. Pathophysiology. 7:153–163. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yin XM and Ding WX: Death receptor
activation-induced hepatocyte apoptosis and liver injury. Curr Mol
Med. 3:491–508. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liu YY, Xie Q, Wang H, Lin LY, Jiang S,
Zhou XQ, Yu H and Guo Q: The effect of N-acetyl-L-cysteine on
endoplasmic reticulum stress mediated apoptosis of HepG2 cells.
Zhonghua Gan Zang Bing Za Zhi. 16:524–527. 2008.(In Chinese).
PubMed/NCBI
|
|
38
|
Loos B, Genade S, Ellis B, Lochner A and
Engelbrecht AM: At the core of survival: autophagy delays the onset
of both apoptotic and necrotic cell death in a model of ischemic
cell injury. Exp Cell Res. 317:1437–1453. 2011. View Article : Google Scholar : PubMed/NCBI
|