Introduction
Neovascularization is an important characteristic
pathology of diabetic retinopathy (DR), retinopathy of prematurity
and other hypoxic-ischemic retinal diseases (1). The resulting ectopic network of blood
vessels can cause hemorrhage in the retina, choroid and vitreous,
leading to retinal detachment and irreversible blindness. These
diseases are one of the main causes of acquired blindness worldwide
(2), and reliable animal models
are required to further understand the underlying molecular
mechanisms in order to develop preventative strategies.
The oxygen-induced retinopathy (OIR) model for
research into retinal neovascularization was first described by
Smith et al (3) in 1994. In
this murine model there are two phases in the OIR pathological
process: A 75% oxygen phase (hyperoxia) and a 21% oxygen phase
(relative hypoxia). Hyperoxia induces the cessation of normal
vessel growth and the regression of existing vessels, accompanied
by a decrease in vascular endothelial growth factor (VEGF)
expression. Relative hypoxia then induces an overexpression of
pro-angiogenic substances, including VEGF, that causes the retina
to initiate pathological neovascularization (3). Due to the pivotal role of VEGF in
this angiogenesis, VEGF may be a potential novel therapeutic target
for retinal neovascular diseases.
β-2-glycoprotein I (β2GPI), also known as
apolipoprotein H, downregulates VEGF signaling pathways and
inhibits angiogenesis (4).
Therefore, β2GPI represents a potential target in the treatment of
DR. β2GPI is a phospholipid-binding plasma protein consisting of
five homologous repeated units (domains I to V) (5). The cysteine (Cys) 288-Cys326
disulfide bond in domain V can be reduced by thiol oxidoreductase
thioredoxin-1 or protein disulfide isomerase to form reduced β2GPI
(6). Reduced β2GPI exhibits
increased binding to von Willebrand factor through disulfide bonds
and is able to protect endothelial cells, including EA.hy926 human
endothelial cells, from oxidative stress-induced cell death in
vitro (7,8). Reduced β2GPI may therefore attenuate
neovascularization through the amelioration of oxidative stress or
through direct effects on VEGF activity.
In the present study, a mouse model of OIR was used
to investigate the effects of β2GPI and reduced β2GPI on retinal
neovascularization and to delineate the underlying molecular
mechanisms.
Materials and methods
Animals and reagents
C57BL/6J newborn mice and nursing female mice were
obtained from the Experimental Animal Center of Peking University
Health Science (Beijing, China). β2GPI and reduced β2GPI were
prepared as previously described (6). Evans Blue, diethylpyrocarbonate, EDTA
and hematoxylin and eosin (H&E) were purchased from
Sigma-Aldrich (St Louis, MO, USA); RNasin Inhibitor and Moloney
Murine Leukemia Virus (M-MLV) Reverse Transcriptase were purchased
from Promega Corp. (Madison, WI, USA); SYBR® Premix Ex
Taq™ DNA polymerase was purchased from Takara (Dalian, China) and
TRIzol™ reagent was purchased from Invitrogen Life Technologies
(Carlsbad, CA, USA). Rabbit anti-mouse polyclonal antibodies to
Akt, phosphorylated- (p-) Akt, extracellular signal-regulated
kinase 1/2 (ERK1/2), p-ERK1/2 and cluster of differentiation (CD)
34 were purchased from Bioworld Technology, Inc. (St. Louis Park,
MN, USA). The quantitative polymerase chain reaction (qPCR)
reaction primer sequences were designed by Primer 5.0 software
(Premier Biosoft International, Palo Alto, CA, USA). and were
synthesized by Beijing Genomics Institute (Beijing, China).
Animal model of proliferative
retinopathy
All applicable institutional (Tianjin Medical
University, Tianjin, China) and governmental regulations concerning
the ethical use of animals were followed during this study. A
reproducible model of OIR has been described previously in detail
(3). Briefly, seven-day-old
C57BL/6J mice, together with their nursing mothers, were exposed to
75±2% oxygen (hyperoxia) for five days before being returned to
normal atmospheric conditions for a further five days. These mice
were then randomly divided into three treatment groups and
administered intravitreal injections of (respectively) 1 μl
phosphate-buffered saline (PBS), 1 μl 800 μg/ml β2GPI or 1 μl 800
μg/ml reduced β2GPI. Mice of the same strain and age were kept in
normal atmospheric conditions for use as a control group.
Histological quantification of retinal
neovascularization and immunohistochemistry
Mice were sacrificed by cervical vertebra
dislocation. The eyes were enucleated, fixed with 4% (w/v)
paraformaldehyde in PBS and embedded in paraffin. Starting at the
optic nerve head, a series of 6-μm, paraffin-embedded axial retinal
sections were obtained, and these sections were stained with
H&E. Sections were examined for evidence of neovascularization
by an individual blinded to the treatment groups using light
microscopy. Neovascularization was quantified by the number of
retinal vascular endothelial cell nuclei anterior to the inner
limiting membrane (ILM). Counts were performed in 10 transverse
sections through the center of the eye to produce an average number
of neovascular cell nuclei/section/eye.
The paraffin sections underwent dewaxing,
rehydration, antigen retrieval and treatment with 4% hydrogen
peroxidase. Normal goat serum was used as a protein block and the
sections were then incubated with rabbit anti-mouse polyclonal
anti-CD34 antibody overnight at 4°C. Following incubation with the
primary antibody, the sections were incubated with biotinylated
rabbit anti-mouse secondary antibody (diluted 1:200; Boster
Biological Technology Co., Wuhan, China) for 20 min at 37°C and
washed three times with PBS. Streptavidin-Horseradish Peroxidase
Conjugate dilution buffer (Boster Biological Technology Co.) was
subsequently applied for 20 min at 37°C prior to washing three
times with PBS. The tissue sections were incubated in
diaminobenzidine solution and counterstained with hematoxylin,
prior to being dehydrated and mounted. PBS, in place of mouse
monoclonal anti-CD34 antibody, was used as a negative control.
Angiography with Evans Blue
Mice were anesthetized by intraperitoneal injection
of sodium pentobarbital (300 mg/kg) and were sacrificed by
intracardiac perfusion with 1 ml normal saline containing 30 mg/ml
Evans Blue. The eyes were subsequently enucleated and fixed in 4%
(w/v) paraformaldehyde for 2–3 h at room temperature. The retinas
were then dissected into four quadrants and flat-mounted onto
microscope slides with neutral resin glue. Images, which were
obtained by fluorescence microscopy, were analyzed using Adobe
Photoshop (Adobe Systems Software Inc., San Jose, CA, USA) and the
areas of non-perfusion and neovessels were calculated as a
percentage of the total area of the retina.
Reverse transcription-qPCR
Enucleated eyes were used to prepare fresh retinas
for RNA isolation. Total RNA was isolated using TRIzol™ reagent
according to the manufacturer’s instructions (Invitrogen Life
Technologies). Having tested for nucleic acid integrity, an aliquot
of RNA extract (1.5 μg) was reverse-transcribed into cDNA using
M-MLV Reverse Transcriptase. Primers were designed using Primer 5.0
software (Premier Biosoft, Palo Alto, CA, USA) and had the
following sequences: VEGF forward, 5′-CTGGGCACTGCCTGGAAGAAT-3′ and
reverse, 5′-GGAAGATGAGGAAGGGTAAGC-3′; VEGFR-1 forward,
5′-CAAGCCAACGTCCAACAGGAT-3′ and reverse,
5′-GCCCAGCAGAGTGCTAGTGTC-3′; VEGFR-2 forward,
5′-CAAGCCAACGTCCAACAGGAT-3′ and reverse,
5′-CCCTGAGTCAGCGTGAACTGC-3′; hypoxia-inducible factor-1 (HIF-1)
forward, 5′-ATAAATGTTCTGCCCACCCTG-3′ and reverse,
5′-GACCCAACCACAAAGAGCAAG-3′; β-actin forward,
5′-TGGAGAAGAGCTATGAGCTGCCTG-3′ and reverse,
5′-GTGCCACCAGACAGCACTGTGTTG-3′. The PCR was perfomed using an iQ™5
Real-Time PCR Detection system (Bio-Rad Laboratories, Hercules, CA,
USA). PCR cycling conditions were set as follows: 95°C for 5 min,
40 cycles at 95°C for 30 sec, then 56°C for 30 sec and 72°C for 30
sec. Melting curve analysis was subsequently performed between 55
and 95°C by monitoring fluorescence with 0.5°C increments at 30-sec
intervals. All sample measurements were performed in triplicate.
Estimates of the quantity of the PCR product were obtained by
densitometry using the Quantity One® analysis software
package (Bio-Rad Laboratories). For each experimental sample,
target mRNA levels were normalized to β-actin mRNA levels.
Western blot analysis
Mouse retinas were collected and incubated in lysis
buffer containing protease inhibitors (20 mM Tris, pH 7.4; 150 mM
NaCl; 1 mM EDTA; 1 mM phenylmethylsulfonyl fluoride; 1 mM
orthovanadate; 1 μg/ml leupeptin and 10 μg/ml aprotinin). Following
homogenization and centrifugation, the amount of protein in the
supernatant was determined with the bicinchoninic acid protein
assay kit (Sigma-Aldrich). Aliquots of extract (35 μg protein per
lane) from each sample were separated by SDS-PAGE using a 10%
Tris-glycine gel prior to being transferred onto a polyvinylidene
fluoride (PVDF) membrane at 65 V for 2 h. Following blocking of the
nonspecific binding sites by incubation with 5% skimmed milk for 1
h, the membrane was incubated with rabbit anti-mouse polyclonal
antibodies against Akt, p-Akt, ERK and p-ERK (1:1,000 dilution)
overnight at 4°C. The membrane was subsequently incubated with
horseradish peroxidase-conjugated secondary antibody (goat
anti-rabbit) for 1 h at room temperature. Peroxidase activity on
the PVDF membranes was visualized on X-ray film with an
ultra-violet transmission analyzer (GE Healthcare, Piscataway, NJ,
USA).
Statistical analysis
Data were analyzed using SPSS 18.0 (SPSS Inc.,
Chicago, IL, USA). The results are expressed as the mean ± standard
error of the mean. One-way analysis of variance was used to
evaluate statistically significant differences. P<0.05 was
considered to indicate a statistically significant difference.
Results
Histological quantification of retinal
neovascularization and immunohistochemistry
The histological quantification and
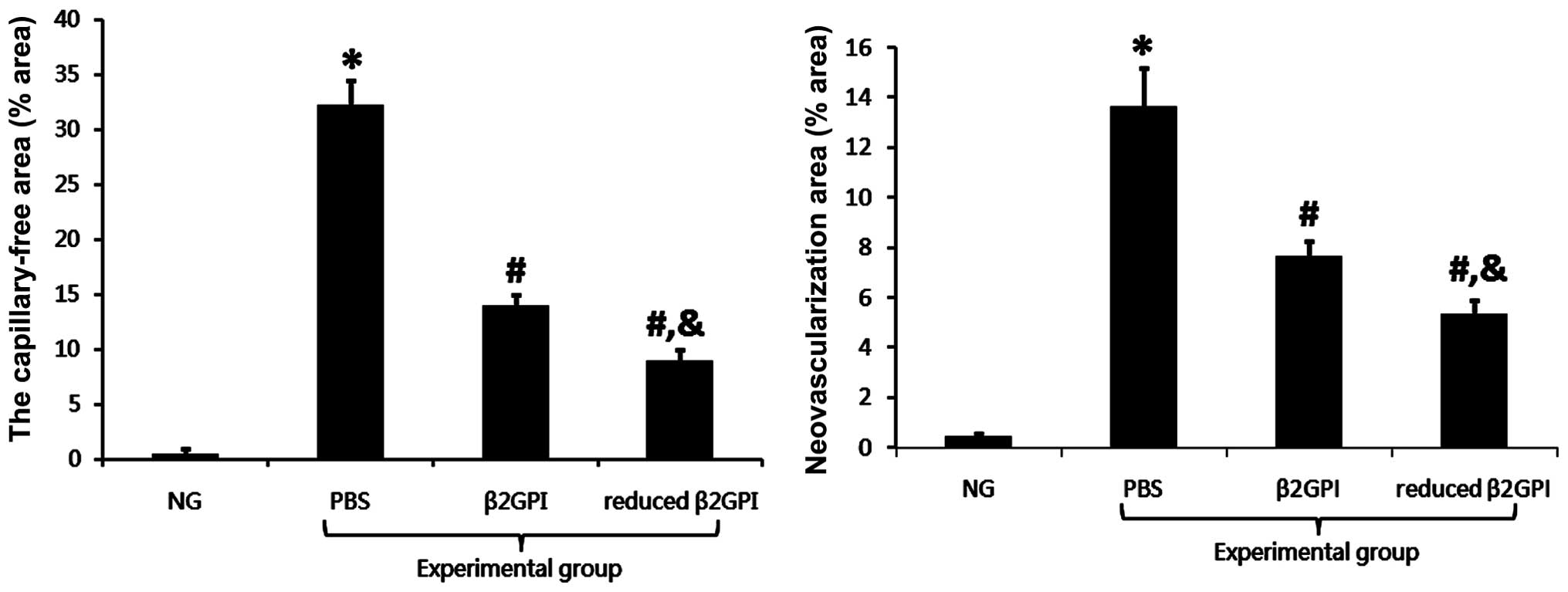
immunohistochemistry results are shown in Fig. 1. The OIR protocol resulted in a
>40-fold increase in the number of endothelial cell nuclei
anterior to the ILM (controls, 0.67±0.816 vs. PBS, 32.5±7.342;
P<0.05). Both β2GPI and reduced β2GPI significantly inhibited
this increase (9.33±2.16 and 5.17±2.32, respectively; P<0.01),
with the effect of the reduced β2GPI observed to be statistically
significantly stronger (P<0.05) (Fig. 1A and C). Immunohistochemical
staining demonstrated that the cells anterior to the ILM were
vascular endothelial cells (Fig.
1B).
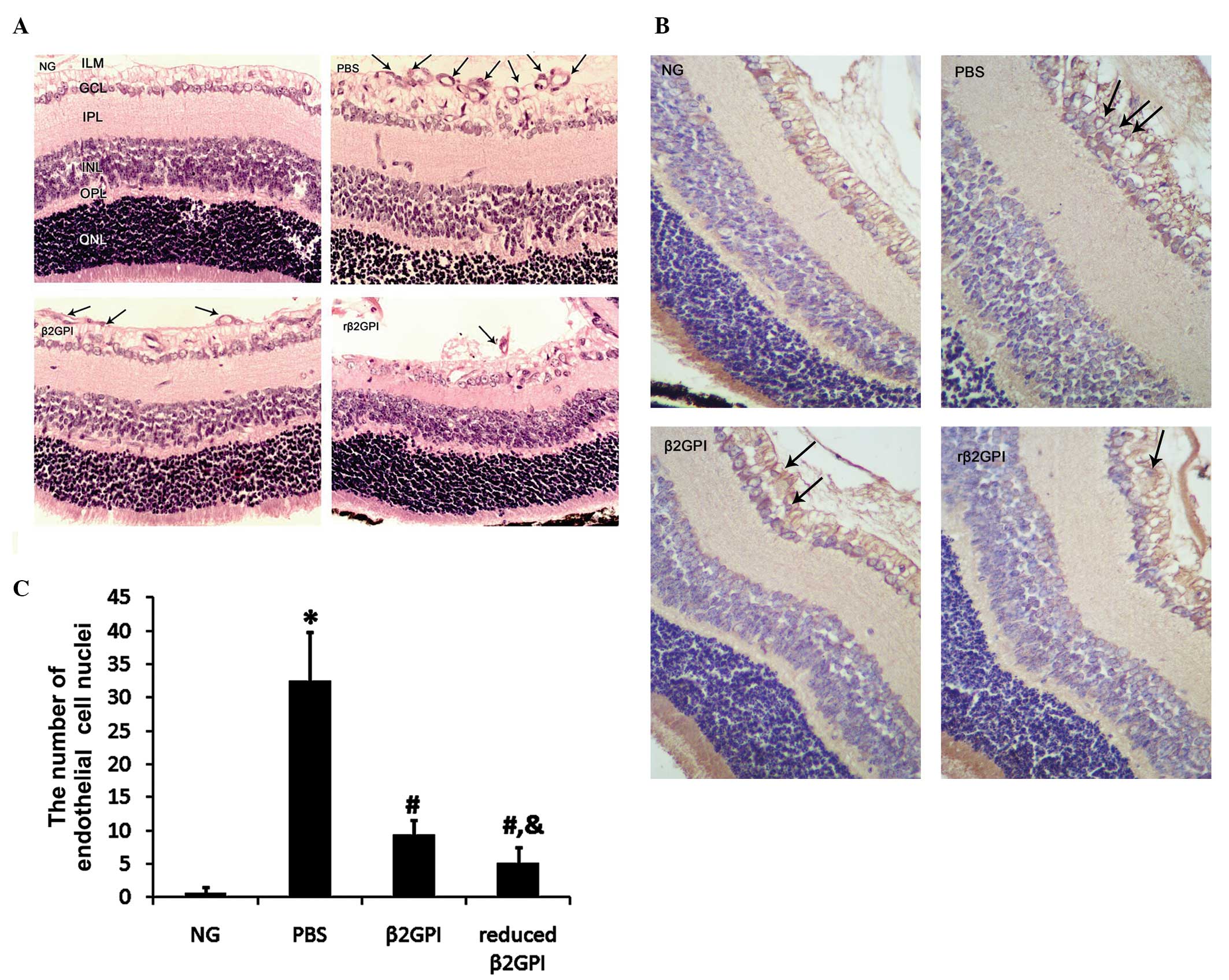 | Figure 1Histological quantification of retinal
neovascularization. Neonatal mice were kept as controls or
subjected to the oxygen-induced retinopathy protocol. (A)
Neovascularization was assessed in hematoxylin and eosin-stained
sections of the retina by counting the number of vascular
endothelial cell nuclei (indicated by arrows) anterior to the ILM.
(B) CD34 immunohistochemical staining of the retina was performed
to reveal angiogenic endothelial cells (brown stain indicated by
arrows). (C) Formal quantification of neovascularization
observations. n=8 in each group. Magnification, ×400. Data are
presented as the mean ± standard error of the mean.
*P<0.01 vs. the NG group; #P<0.01 vs.
the PBS group; &P<0.05 vs. the β2GPI group. NG,
neonatal controls; PBS, phosphate-buffered saline; β2GPI,
β-2-glycoprotein I; rβ2GPI, reduced β2GPI; ILM, inner limiting
membrane; GCL, ganglion cell layer; IPL, inner plexiform layer;
INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer
nuclear layer; CD, cluster of differentiation. |
Effects of β2GPI and reduced β2GPI on
neovessels in the OIR mouse model
The vessels in the normal control group were mature
and showed a normal radial distribution from the optic disc, as
well as exhibiting a thicker diameter and a clear peripheral
retinal structure, with the vessels forming vascular arcades along
the peripheral retina. However, in the OIR PBS group, the diameter
of blood vessels was reduced, and there was evidence of retinal
neovessel formation, vessel leakage and an enlargement of the
non-perfusion region. When formally quantified, the differences
between the PBS and control groups were found to be statistically
significant (P<0.01). Treatment with β2GPI resulted in
restoration of vessel thickness. Furthermore, the formation of
retinal neovessels, the vessel leakage and the enlarged
non-perfusion region were significantly reduced as compared with
the PBS group (P<0.01). This abrogation was even more marked
with reduced β2GPI treatment (Fig.
2).
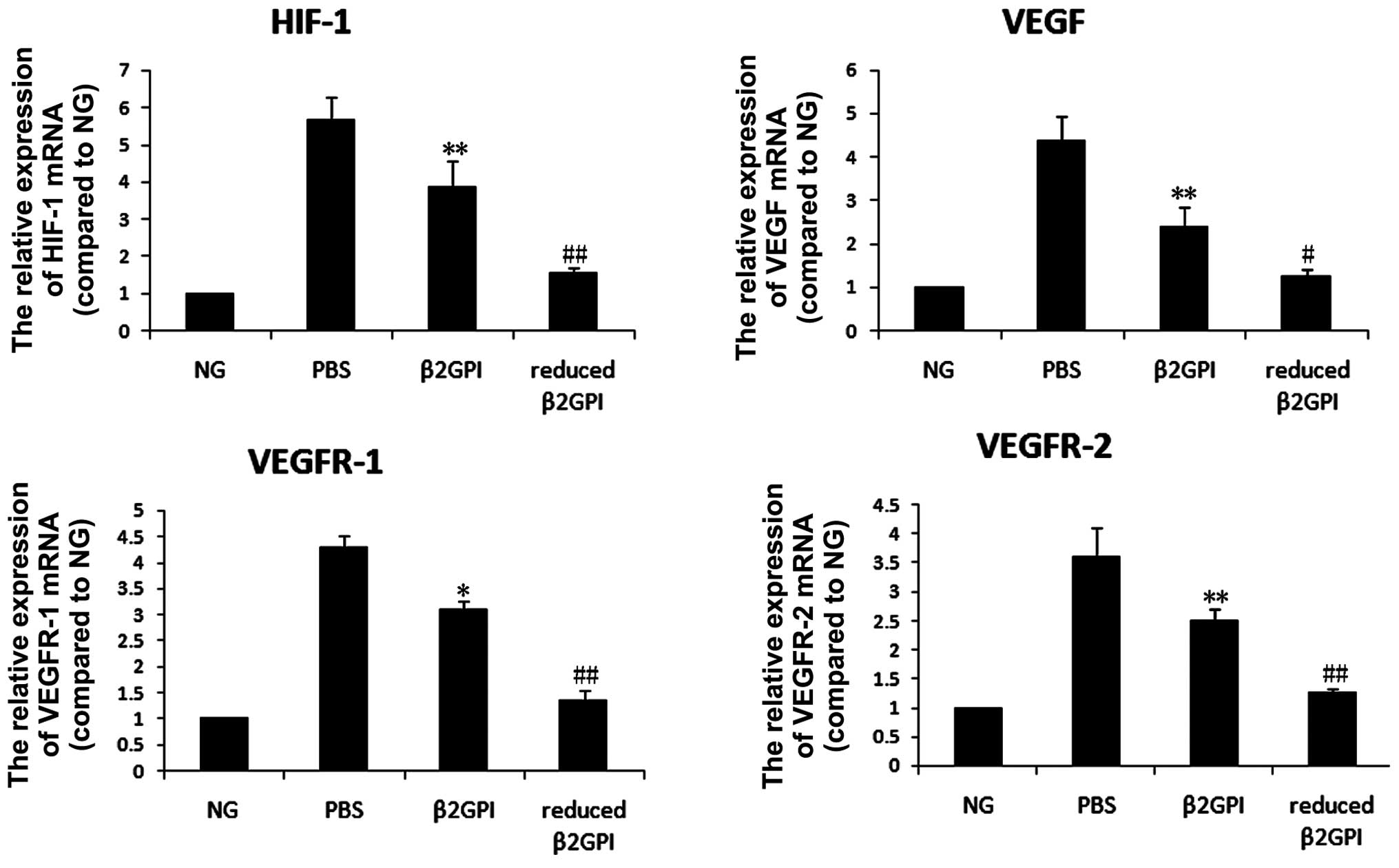
Effects of β2GPI and reduced β2GPI on
mRNA expression in retinopathy
The mRNA expression levels of VEGF, VEGFR-1, VEGFR-2
and HIF-1 were increased 2.5–4-fold by the induction of retinopathy
(P<0.05 or P<0.01) in the PBS group compared with the levels
in the untreated control group. β2GPI and reduced β2GPI both
inhibited this increase (P<0.05 and P<0.01 respectively),
with the effect of reduced β2GPI observed to be stronger than that
of β2GPI (P<0.05 or P<0.01) (Fig. 3).
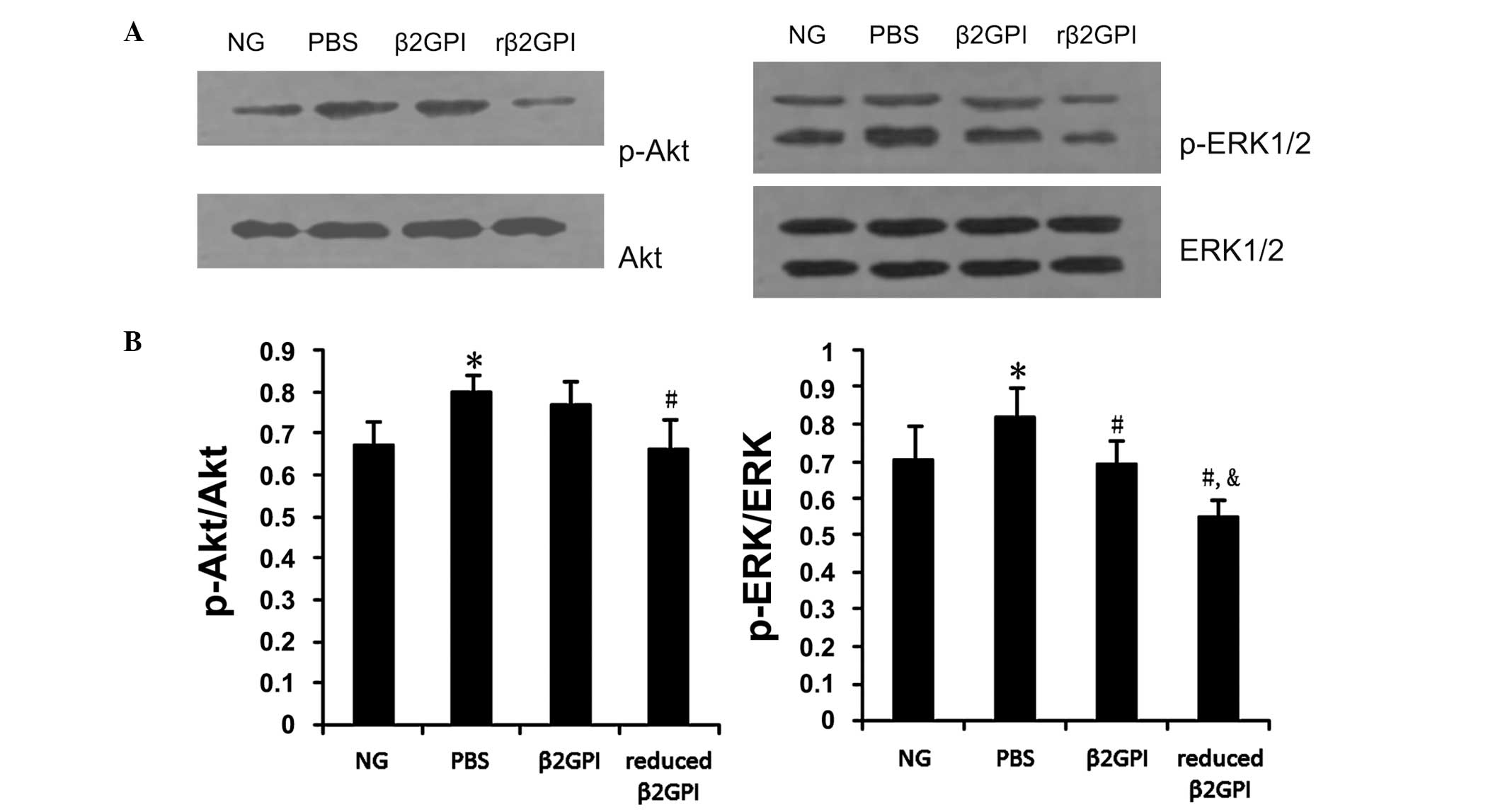
Effects of β2GPI and reduced β2GPI on the
VEGF signaling pathway
Statistically significant increases in p-ERK1/2 and
p-Akt levels were observed in the PBS-treated group (P<0.05), as
compared with the untreated control group. β2GPI inhibited this
increase in p-ERK1/2 in the retina (P<0.05); however, although
the expression of p-Akt decreased following treatment with β2GPI,
this did not attain statistical significance. The expression of
p-Akt and p-ERK1/2 in the retina showed a statistically significant
decrease upon treatment with reduced β2GPI (P<0.05) when
compared with the expression in the PBS and β2GPI groups (Fig. 4).
Discussion
Proliferative DR (PDR) is one of the most severe
microvascular complications in patients with diabetes and is the
major cause of acquired blindness. One important pathogenic
mechanism in PDR is neovascularization through inappropriate
angiogenesis. Passam et al (9) showed that the plasma glycoprotein
β2GPI has the potential to reduce angiogenesis; through the same
mechanism, β2GPI reduces the growth of tumor implants in mouse
models (10). In the present
study, it was shown that β2GPI exerts a stronger effect than β2GPI
in its inhibition of neovascularization. These effects were
regulated through VEGF and its important downstream targets, the
phosphatidylinositol 3-kinase (PI3K)/Akt and mitogen-activated
protein kinase (MAPK)/ERK signaling pathways. Angiogenesis is a
complicated physiological process in vivo. In addition to
angiogenic factors, such as VEGF, fibroblast growth factor and
angiopoietin (11–14), this process includes
anti-angiogenic factors: Platelet factor-4, the expression of which
can be increased by β2GPI (15),
angiostatin, thrombospondin-1 and numerous others (16–19).
Angiogenic and anti-angiogenic factors maintain a dynamic balance
under physiological conditions. A disruption of this balance can
result in various pathologies. When anti-angiogenic factors
dominate, the blood vessels are in a stationary or degradative
state; when angiogenic factors dominate, the blood vessel formation
process is initiated.
In this study, it was shown that reduced β2GPI
inhibited retinal angiogenesis. This was accompanied by the
downregulation of VEGF, a key pro-angiogenic factor, and VEGFR-1/2.
It remains unclear how reduced β2GPI downregulates VEGF and VEGFR;
however, the data presented in this study showed that reduced β2GPI
downregulates the expression of VEGF and VEGFR at the
transcriptional level.
The PI3K/Akt signaling pathway influences
angiogenesis and nutrient supply through the modulation of various
factors, including endothelial nitric oxide synthase, HIF-1 and
glycogen synthase kinase 3 (20,21),
and promotes a survival response in endothelial cells, thus
preventing apoptosis of these cells. This signaling pathway is also
involved in preventing the degradation of newly-formed, immature
lumen. Data from this study showed that reduced β2GPI significantly
inhibited VEGF-induced Akt phosphorylation. ERK, a central protein
of the MAPK/ERK signaling pathway (22), plays a key role in angiogenesis by
entering the nucleus and activating nuclear factor
κ-light-chain-enhancer of activated B cells (23,24),
followed by the induction of DNA synthesis and cell proliferation.
The findings in the present study showed that reduced β2GPI blocked
the phosphorylation of ERK1/2, thus suggesting this mechanism to be
causative for the negative effect of reduced β2GPI on retinal
angiogenesis.
In conclusion, β2GPI and reduced β2GPI may have
potential anti-angiogenic activity in vivo. Both
significantly inhibited pathological retinal angiogenesis, with the
effect of reduced β2GPI observed to be stronger. β2GPI and reduced
β2GPI were shown to act by downregulating the expression of HIF-1,
VEGF and its receptors, VEGFR-1/VEGFR-2, on endothelial cells, and
blocking the phosphorylation of ERK1/2 and Akt, downstream targets
of VEGF in the MAPK/ERK and PI3K/Akt pathways. Intravitreal
injection of reduced β2GPI to inhibit neovascularization may
therefore provide a novel approach to the treatment of PDR.
Acknowledgements
The authors would like to thank the National Natural
Science Foundation of China (nos. 30971393 and 81070645), the
Tianjin Natural Science Fund (nos. 10JCYBJC12000 and
13ZCZDSYO1300), the Tianjin Health Bureau Natural Science Fund
(nos. 09KZ01, 09KZ89 and 12KG135) and the Tianjin Medical
University Science and Technology Fund (no. 2009ky25).
References
|
1
|
Tolentino MJ: Current molecular
understanding and future treatment strategies for pathologic ocular
neovascularization. Curr Mol Med. 9:973–981. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang FH, Liang YB, Zhang F, et al:
Prevalence of diabetic retinopathy in rural China: the Handan Eye
Study. Ophthalmology. 116:461–467. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Smith LE, Wesolowski E, McLellan A, et al:
Oxygen-induced retinopathy in the mouse. Invest Ophthalmol Vis Sci.
35:101–111. 1994.PubMed/NCBI
|
|
4
|
Yu P, Passam FH, Yu DM, Denyer G and
Krilis SA: Beta2-glycoprotein I inhibits vascular endothelial
growth factor and basic fibroblast growth factor induced
angiogenesis through its amino terminal domain. J Thromb Haemost.
6:1215–1223. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
de Laat B, van Berkel M, Urbanus RT, et
al: Immune responses against domain I of β(2)-glycoprotein I are
driven by conformational changes: domain I of β(2)-glycoprotein I
harbors a cryptic immunogenic epitope. Arthritis Rheum.
63:3960–3968. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Passam FH, Rahgozar S, Qi M, et al: Beta 2
glycoprotein I is a substrate of thiol oxidoreductases. Blood.
116:1995–1997. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Passam FH, Rahgozar S, Qi M, et al: Redox
control of β2-glycoprotein I-von Willebrand factor interaction by
thioredoxin-1. J Thromb Haemost. 8:1754–1762. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ioannou Y, Zhang JY, Passam FH, et al:
Naturally occurring free thiols within beta 2-glycoprotein I in
vivo: nitrosylation, redox modification by endothelial cells and
regulation of oxidative stress induced cell injury. Blood.
116:1961–1970. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Passam FH, Qi JC, Tanaka K, Matthaei KI
and Krilis SA: In vivo modulation of angiogenesis by beta 2
glycoprotein I. J Autoimmun. 35:232–240. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Beecken WD, Engl T, Ringel EM, et al: An
endogenous inhibitor of angiogenesis derived from a transitional
cell carcinoma: clipped beta2-glycoprotein-I. Ann Surg Onco.
13:1241–1251. 2006. View Article : Google Scholar
|
|
11
|
Yoncopoulos GD, Davis S, Gale NW, Rudge
JS, Wiegand SJ and Holash J: Vascular-specific growth factors and
blood vessel formation. Nature. 407:242–248. 2000. View Article : Google Scholar
|
|
12
|
Yoshida S, Yoshida A and Ishibashi T:
Induction of IL-8, MCP-l, and bFGF by TNF-alpha in retinal glial
cells: implications for retinal neovascularization during
post-ischemic inflammation. Graefes Arch Clin Exp Ophthalmol.
242:409–413. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Feng Y, vom Hagen F, Pfister F, et al:
Impaired pericyte recruitment and abnormal retinal angiogenesis as
a result of angiopoietin-2 overexpression. Thromb Haemost.
97:99–108. 2007.PubMed/NCBI
|
|
14
|
Gardiner TA, Gibson DS, de Gooyer TE, de
la Cruz VF, McDonald DM and Stitt AW: Inhibition of tumor necrosis
factor-alpha improves physiological angiogenesis and reduces
pathological neovascularization in ischemic retinopathy. Am J
Pathol. 166:637–644. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang R, Zhou SJ, Li CJ, et al: C-reactive
protein/oxidised low-density lipoprotein/β2-glycoprotein I complex
promotes atherosclerosis in diabetic BALB/c mice via
p38mitogen-activated protein kinase signal pathway. Lipids Health
Dis. 12:422013. View Article : Google Scholar
|
|
16
|
Ljubimov AV, Caballero S, Aoki AM, Pinna
LA, Grant MB and Castellon R: Involvement of protein kinase CK2 in
angiogenesis and retinal neovascularization. Invest Ophthalmol Vis
Sci. 45:4583–4591. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Uusitalo-Jarvinen H, Kurokawa T, Mueller
BM, Andrade-Gordon P, Friedlander M and Ruf W: Role of protease
activated receptor l and 2 signaling in hypoxia-induced
angiogenesis. Arterioscler Thromb Vasc Biol. 27:1456–1462. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Qiao H, Sonoda KH, Ikeda Y, et al:
Interleukin-18 regulates pathological intraocular
neovascularization. J Leukoc Biol. 81:1012–1021. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu Z, Wang S, Sorenson CM and Sheibani N:
Attenuation of retinal vascular development and neovascularization
in transgenic mice over-expressing thrombospondin-l in the lens.
Dev Dyn. 235:1908–1920. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tchaikovski V, Olieslagers S, Böhmer FD
and Waltenberger J: Diabetes mellitus activates signal transduction
pathways resulting in vascular endothelial growth factor resistance
of human monocytes. Circulation. 120:150–159. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Park JH, Lee JY, Shin DH, Jang KS, Kim HJ
and Kong G: Loss of Mel-18 induces tumor angiogenesis through
enhancing the activity and expression of HIF-1α mediated by the
PTEN/PI3K/Akt pathway. Oncogene. 30:4578–4589. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Modi H, Li L, Chu S, Rossi J, Yee JK and
Bhatia R: Inhibition of Grb2 expression demonstrates an important
role in BCR-ABL-mediated MAPK activation and transformation of
primary human hematopoietic cells. Leukemia. 25:305–312. 2011.
View Article : Google Scholar :
|
|
23
|
Parikh N, Shuck RL, Nguyen TA, Herron A
and Donehower LA: Mouse tissues that undergo neoplastic progression
after K-Ras activation are distinguished by nuclear translocation
of phospho-Erk1/2 and robust tumor suppressor responses. Mol Cancer
Res. 10:845–855. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Taylor SM, Nevis KR, Park HL, et al:
Angiogenic factor signaling regulates centrosome duplication in
endothelial cells of developing blood vessels. Blood.
116:3108–3117. 2010. View Article : Google Scholar : PubMed/NCBI
|