Introduction
The global lung cancer mortality rate is the highest
among all types of cancer and its incidence is gradually increasing
(1). Non-small cell lung cancer
(NSCLC) is the most common type of lung cancer (accounting for 80%
of all cases), and includes squamous cell carcinoma, adenocarcinoma
and large cell carcinoma. Although surgical resection, radiation
therapy and chemotherapy technology continue to improve gradually,
patients with lung cancer remain exceedingly vulnerable to relapse
and mortality (2). The global cure
rate of lung cancer is low and the average 5-year survival rate is
<15% (3–6). However, the mechanisms of NSCLC have
not been elucidated, and hence the study of NSCLC is crucial.
Long-chain non-coding RNAs (long non-coding RNAs,
lncRNAs) are RNA molecules with a transcript longer than 200
nucleotides in the nucleus or cytoplasm (7). LncRNAs are usually divided into five
categories: Sense, antisense, bidirectional, introns and intergenic
lncRNAs. In recent years, a large number of lncRNAs have been
identified and a human lncRNA database providing details of lncRNA
expression and other significant information has been established
(8). Numerous studies have linked
the lncRNAs with diseases, and abnormal expression has been noted
in a range of diseases, including cancer (9,10).
Studies have demonstrated that lncRNAs are
differentially expressed in normal cells and tumor cells, and since
lncRNAs are a significant regulatory factor of gene expression,
their aberrant expression will inevitably lead to abnormalities in
gene expression and tumorigenesis. LncRNA disorders are also a
feature of several types of cancer and promote the development,
invasion and metastasis of tumors by a variety of mechanisms
(9,11). LncRNAs regulate the transcriptional
expression at the epigenetic, transcription and post-transcription
levels (12–14).
Previous studies have demonstrated that lncRNAs are
involved in the development and progression of NSCLC. However,
research into lncRNAs in NSCLC is in its infancy and only a small
number of NSCLC-associated lncRNAs have been identified, including
lcRNA HOTAIR, lcRNA H19, lcRNA ANRIL, lcRNA MALAT1 (15,16)
and lcRNA SCAL1 (17), lncRNA
AK126698 (18) and lncRNA GAS6-AS1
(19). However, lncRNAs of NSCLC
require further study to elucidate their mechanism of action.
In this study, we detected the lncRNA and mRNA
expression patterns in NSCLC samples compared with corresponding
adjacent normal tissue (NT) samples, several of which were
evaluated by reverse transcription-quantitative polymerase chain
reaction (RT-qPCR) in a total of 90 pairs of tissues. The results
revealed that lncRNA expression patterns may provide new molecular
biomarkers for the diagnosis of NSCLC.
Materials and methods
Patient samples
NSCLC and corresponding NT samples were
prospectively collected from 105 patients at The First Affiliated
Hospital of Wenzhou Medical University, China, from April 2012 to
August 2013. Samples from 15 of the patients were used for
microarray analysis of lncRNAs and those from the remaining 90 were
used for additional evaluations (Table
I). The diagnosis of adenocarcinoma was confirmed by the
histopathological results. The NSCLC and matched NT samples were
snap-frozen in liquid nitrogen immediately after resection. The
study was approved by the Institutional Ethics Review Board of The
First Affiliated Hospital of Wenzhou Medical University, and all
patients provided written informed consent for this study.
 | Table IDemographical characteristics of 90
cases of non-small cell lung cancer. |
Table I
Demographical characteristics of 90
cases of non-small cell lung cancer.
| Parameter | Year/number |
|---|
| Age (years) | 64.5 (37–80) |
| Gender
(female/male) | 50/40 |
| Histological
grade |
| Well-differentiated
carcinoma | 13 |
| Well- to moderately
differentiated carcinoma | 15 |
| Moderately
differentiated carcinoma | 32 |
| Moderately to poorly
differentiated carcinoma | 12 |
| Poorly
differentiated carcinoma | 18 |
| TNM clinical
stagea |
| Ia | 22 |
| Ib | 36 |
| IIa | 11 |
| IIb | 5 |
| IIIa | 16 |
RNA extraction
NSCLC cells were obtained by laser microdissection;
the proportion of cancer cells in the tissue sections was 100%. The
15 NSCLC specimens were divided into three groups; namely, every
five samples from NSCLC were combined into a group. Next, 15 of the
corresponding NT samples were mixed into one group. The four groups
were subjected to RNA extraction. Total RNA was extracted using
TRIzol reagent (Invitrogen Life Technologies, Carlsbad, CA, USA),
according to the manufacturer’s instructions. The integrity of the
RNA was assessed by electrophoresis on a denaturing agarose gel. An
ND-1000 spectrophotometer (NanoDrop Technologies, Inc., Wilmington,
DE, USA) was used for the accurate measurement of RNA concentration
(OD260), protein contamination
(OD260/OD280 ratio) and organic compound
contamination (OD260/OD230 ratio).
Microarray and computational
analysis
An Agilent array platform (Agilent Technologies,
Inc., Santa Clara, CA, USA) was employed for microarray analysis.
The sample preparation and microarray hybridization were performed
according to the manufacturer’s instructions with minor
modifications. Briefly, mRNA was purified from total RNA following
the removal of rRNA using an mRNA-ONLY™ eukaryotic mRNA isolation
kit (Epicentre Biotechnologies, Madison, WI, USA). Subsequently,
each sample was amplified and transcribed into fluorescent cRNA
along the entire length of the transcripts without 3′ bias using a
random priming method. The labeled cRNAs were hybridized onto a
Human lncRNA Array v3.0 (8×60 K; Arraystar, Rockville, MD, USA),
designed for 30,586 lncRNAs and 26,109 coding transcripts. The
lncRNAs were carefully constructed using the most highly respected
public transcriptome databases, including Refseq (http://www.ncbi.nlm.nih.gov/refseq/), UCSC Known
Genes (http://www.biomedsearch.com/nih/UCSC-Known-Genes/16500937.html)
and GENCODE (http://www.gencodegenes.org/) as well as landmark
publications (20–22). Each transcript was accurately
identified by a specific exon or splice junction probe. Positive
probes for housekeeping genes and negative probes were also printed
onto the array for hybridization quality control. After washing the
slides, the arrays were scanned using the G2505C scanner (Agilent
Technologies, Inc.), and the acquired array images were analyzed
with the Feature Extraction software (version 11.0.1.1, Agilent
Technologies, Inc.). Quantile normalization and subsequent data
processing were performed using the GeneSpring GX v12.0 software
package (Agilent Technologies, Inc.). The microarray was performed
by KangChen Bio-tech, Shanghai, China.
Functional group analysis
Gene ontology (GO) analysis was derived from Gene
Ontology (www.geneontology.org), which provides
three structured networks of defined terms that describe gene
product attributes. The P-value denotes the significance of GO term
enrichment in the differentially expressed mRNA list (P≤0.05 was
considered to indicate a statistically significant difference).
Pathway analysis was also carried out for the differentially
expressed mRNAs based on the latest Kyoto Encyclopedia of Genes and
Genomes (KEGG; http://www.genome.jp/kegg/) database. This analysis
allowed us to determine the biological pathway for which a
significant enrichment of differentially expressed mRNAs
existed.
RT-qPCR
Total RNA was extracted from frozen NSCLC tissues
with TRIzol reagent (Invitrogen Life Technologies) and then reverse
transcribed using an RT reagent kit (Thermo Fisher Scientific,
Waltham, MA, USA) according to the manufacturer’s instructions.
LncRNA expression in NSCLC tissues was measured by qPCR using SYBR
Premix Ex Taq (Thermo Fisher Scientific) with an ABI 7000
instrument (Applied Biosystems, Inc., Foster City, NJ, USA). Two
lncRNAs that were significantly expressed (RP11-385J1.2 and TUBA4B)
were evaluated in all of the patients included in this study. Total
RNA (2 mg) was transcribed to cDNA. PCR was performed in a total
reaction volume of 20 μl, including 10 μl SYBR Premix (2X), 2 μl
cDNA template, 1 μl PCR forward primer (10 mM;
(5′-TGTCAGACTCTCGGGACCAT-3′ for RP11-385J1.2 and
5′-AAAGTGCAACGTGCCATGTG-3′ for TUBA4B), 1 μl PCR reverse primer (10
mM; 5′-GATGCCACTGGAGTGTTGGA-3′ for RP11-385J1.2 and
5′-CTCCACACTATCCATGCCCA-3′ for TUBA4B) and 6 μl double-distilled
water. The qPCR reaction was performed with an initial denaturation
step of 10 min at 95°C, then 95°C (5 sec) and 60°C (30 sec) for a
total of 40 cycles, with a final extension step at 72°C for 5 min.
All experiments were performed in triplicate and all samples were
normalized to GAPDH. The median in each triplicate was used to
calculate the relative lncRNA concentrations (ΔCt = Ct median
lncRNAs - Ct median GAPDH). The fold changes in expression were
calculated (23).
Statistical methods
The Shapiro-Wilk test was used to evaluate the
distribution. Comparisons between two groups were tested using the
Mann-Whitney U test for non-normal distribution. The fold change
and Student’s t-test were analyzed for statistical significance of
the microarray results. The false discovery rate was calculated to
correct the P-value. The threshold value used to designate
differentially expressed lncRNAs and mRNAs was a fold change of
≥2.0 or ≤0.5. P<0.05 was considered to indicate a statistically
significant difference. SPSS version 18.0 (SPSS Inc., Chicago, IL,
USA) was used for statistical analysis.
Results
Overview of lncRNA expression
profiles
To study the potential biological functions of
lncRNAs in NSCLC, we examined the lncRNA and mRNA expression
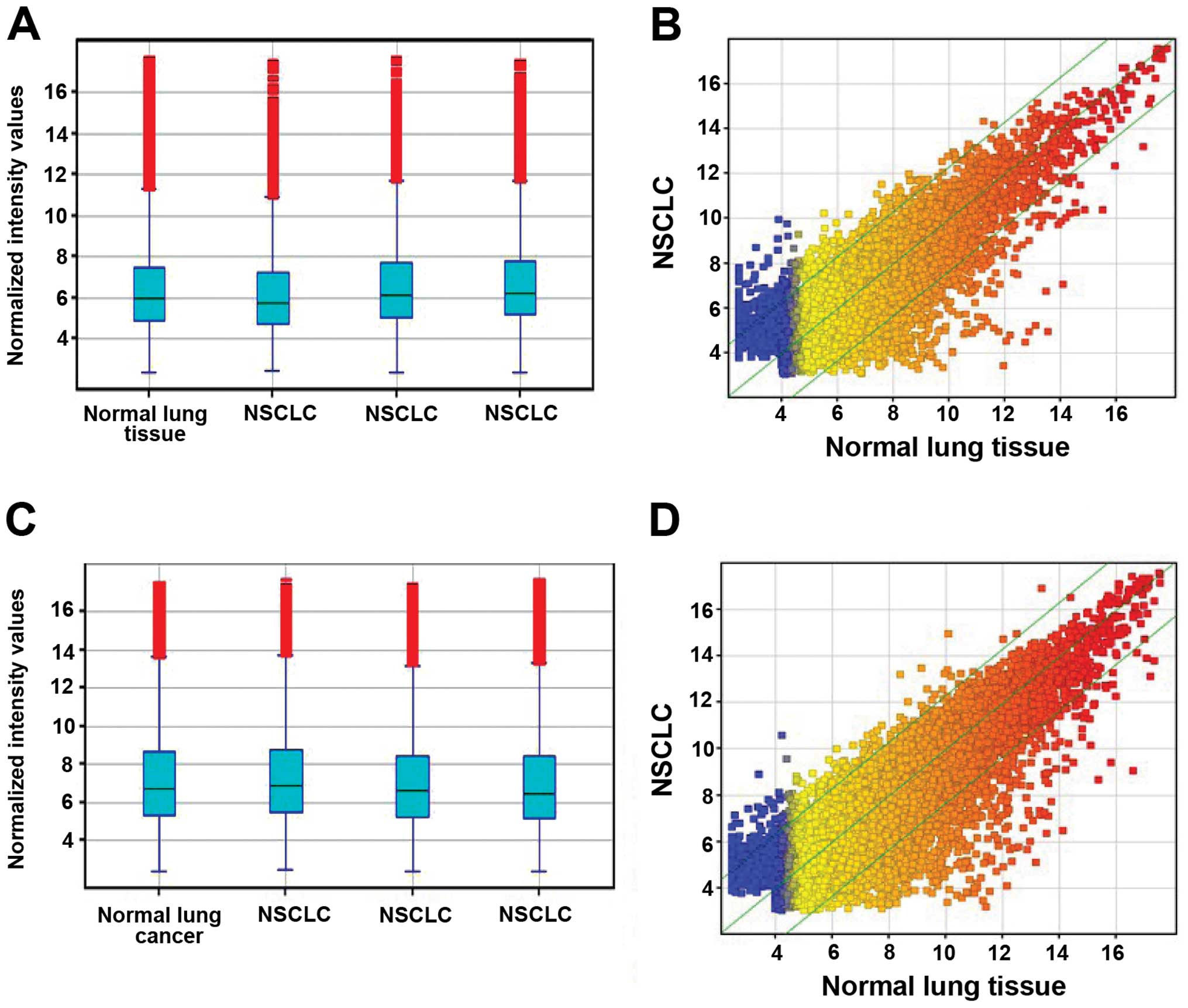
profiles in human NSCLC using microarray analysis (Fig. 1). For this analysis, authoritative
data sources containing >30,586 lncRNAs were used. The
expression profiles of 1,242 lncRNAs indicated that they were
differentially expressed (fold change ≥2.0 or ≤0.5; P<0.05)
between NSCLC and normal lung samples. Among these, 541 lncRNAs
were observed to be upregulated >2-fold in the NSCLC group
compared with the normal lung group, while 701 lncRNAs were
downregulated >2-fold (P<0.05; Table II, Fig. 1A and B, Fig. 2A).
| Table IIUpregulated and downregulated
long-chain non-coding RNAs in non-small cell lung cancer. |
Table II
Upregulated and downregulated
long-chain non-coding RNAs in non-small cell lung cancer.
| Probe name | FC (abs) (NSCLC vs
normal lung tissue) | Regulation | Gene symbol |
|---|
| ASHGA5P051906 | 2.3496380 | Up | RP11-412P11.1 |
| ASHGA5P050658 | 3.3447275 | Up | AC140481.7 |
| ASHGA5P045969 | 2.3421350 | Up | AK129672 |
| ASHGA5P037374 | 3.0890374 | Up | AP001469.9 |
| ASHGA5P027700 | 2.3656695 | Up | FLJ31485 |
| ASHGA5P020784 | 5.6349115 | Up | RP11-909N17.3 |
| ASHGA5P035023 | 2.8399296 | Up | XLOC_002399 |
| ASHGA5P055971 | −3.3054008 | Down | XLOC_012542 |
| ASHGA5P047263 | −3.5518806 | Down | RP11-445K13.2 |
| ASHGA5P031003 | −4.0572240 | Down | CTA-363E6.2 |
| ASHGA5P039685 | −2.9653406 | Down | HSP90AA6P |
| ASHGA5P026985 | −2.3325500 | Down | RP11-264F23.3 |
| ASHGA5P055824 | −4.2268586 | Down | GPC5-AS1 |
| ASHGA5P040177 | −2.7432818 | Down | BX004987.5 |
LncRNA classification and subgroup
analysis
The expression profiles of 343 intergenic lncRNAs
indicated that they were differentially expressed (fold change
≥2.0, P<0.05) between NSCLC and normal lung samples. Among
these, 167 were upregulated and 176 were downregulated. Nearby
coding genes that may be regulated by these lncRNAs were also
identified (Table III). LncRNAs
with enhancer-like function (lncRNA-a) were identified using
GENCODE annotation. The expression profiles of 18 enhancer-like
lncRNAs indicated that they were differentially expressed (fold
change ≥2.0, P<0.05) between NSCLC and normal lung samples.
Among these, seven were upregulated and 11 were downregulated.
Nearby coding genes that may be regulated by these enhancer-like
lncRNAs were also identified (Table
IV). Hox lncRNAs (lncRNAs transcribed from Hox loci lncRNAs)
profiles: This table contains 83 HoxlncRNA clusters (data not
shown).
| Table IIIUpregulated and downregulated
long-chain non-coding RNAs (lncRNAs) in non-small cell lung cancer
and nearby encoding genes regulated by lncRNAs. |
Table III
Upregulated and downregulated
long-chain non-coding RNAs (lncRNAs) in non-small cell lung cancer
and nearby encoding genes regulated by lncRNAs.
| Seqname | Gene symbol | Absolute fold
change lncRNAs | Regulatory
lncRNAs | Nearby gene |
|---|
|
ENST00000562902 | RP11-426C22.5 | −4.2223506 | Down | NM_007245 |
|
ENST00000563624 | RP11-68I18.10 | 2.4656520 | Up | NM_003557 |
|
ENST00000563624 | RP11-68I18.10 | 2.4656520 | Up | NM_207171 |
|
ENST00000564524 | FAM157C | 2.4693956 | Up |
ENST00000555147 |
|
ENST00000564854 | RP13-514E23.1 | −3.2555604 | Down | NM_138980 |
|
ENST00000565118 | ABCC6P1 | −2.4422970 | Down | NM_015161 |
|
ENST00000565153 | RP11-297L17.2 | 2.5062720 | Up | NM_001080430 |
|
ENST00000565862 | RP11-594N15.3 | −2.6900120 | Down |
ENST00000263851 |
|
ENST00000566420 | RP11-506E9.3 | −3.4461820 | Down | NM_153699 |
|
ENST00000566942 | RP11-284N8.3 | −3.9437150 | Down | NM_001040033 |
| Table IVEnhancer-like long-chain non-coding
RNAs (lncRNAs) in non-small cell lung cancer and nearby encoding
genes regulated by lncRNAs. |
Table IV
Enhancer-like long-chain non-coding
RNAs (lncRNAs) in non-small cell lung cancer and nearby encoding
genes regulated by lncRNAs.
| Seqname | Gene symbol | Absolute fold
change lncRNAs | Regulatory
lncRNAs | Nearby gene |
|---|
|
ENST00000366140 | AC017076.5 | −2.3342237 | Down | NM_207315 |
|
ENST00000418076 | RP11-37E23.5 | −2.8438077 | Down | NM_001079691 |
|
ENST00000421619 | RP11-114B7.6 | 3.0627985 | Up |
ENST00000373484 |
|
ENST00000421619 | RP11-114B7.6 | 3.0627985 | Up | NM_001145720 |
|
ENST00000428508 | RP11-353N4.1 | −2.7596264 | Down | NM_001123375 |
|
ENST00000433986 | RP11-261C10.3 | 2.8433535 | Up | NM_014812 |
|
ENST00000446476 | RP5-826L7.1 | −3.6936464 | Down | NM_152410 |
|
ENST00000446476 | RP5-826L7.1 | −3.6936464 | Down | NM_206853 |
|
ENST00000446476 | RP5-826L7.1 | −3.6936464 | Down | NM_206854 |
|
ENST00000453853 | RP11-342C24.8 | 2.8042965 | Up |
ENST00000374325 |
Overview of mRNA expression profiles
In total, 1,102 mRNAs were noted to be
differentially expressed between NSCLC and normal lung samples,
including 271 upregulated mRNAs and 831 downregulated mRNAs
(Fig. 1C and D, Fig. 2B).
GO analysis
The genes corresponding to the downregulated mRNAs
included 278 genes involved in biological processes, 75 genes
involved in cellular components and 59 genes involved in molecular
functions. The genes corresponding to the upregulated mRNAs
included 246 genes involved in biological processes, 58 genes
involved in cellular components and 66 genes involved in molecular
functions.
Pathway analysis
Eleven upregulated pathways were identified,
including ethanol metabolism, systemic lupus erythematosus,
transcriptional misregulation in cancer and cell cycle pathways
(Table V). Twenty-eight
downregulated pathways were identified, including malaria, African
trypanosomiasis and allograft rejection (Table VI).
| Table VPathway analysis of upregulated mRNA
in non-small cell lung cancer. |
Table V
Pathway analysis of upregulated mRNA
in non-small cell lung cancer.
| Pathway ID | Definition | Fisher P-value | False discovery
rate | Enrichment
score |
|---|
| hsa05034 | Alcoholism -
Homo sapiens (human) | 7.52732E-07 | 0.000197968 | 6.123360 |
| hsa05322 | Systemic lupus
erythematosus - Homo sapiens (human) | 5.81856E-05 | 0.007651410 | 4.235184 |
| hsa05202 | Transcriptional
misregulation in cancer - Homo sapiens (human) | 0.000123941 | 0.010865460 | 3.906786 |
| hsa04110 | Cell cycle -
Homo sapiens (human) | 0.003491957 | 0.229596200 | 2.456931 |
| hsa04070 |
Phosphatidylinositol signaling system -
Homo sapiens (human) | 0.009181137 | 0.466312700 | 2.037104 |
| hsa04744 | Phototransduction -
Homo sapiens (human) | 0.010638310 | 0.466312700 | 1.973127 |
| hsa05214 | Glioma - Homo
sapiens (human) | 0.019614550 | 0.678238400 | 1.707422 |
| hsa05212 | Pancreatic cancer -
Homo sapiens (human) | 0.020630820 | 0.678238400 | 1.685483 |
| hsa04114 | Oocyte meiosis -
Homo sapiens (human) | 0.032740740 | 0.956757000 | 1.484912 |
| hsa05203 | Viral
carcinogenesis - Homo sapiens (human) | 0.046635330 | 1 | 1.331285 |
| hsa04512 | ECM-receptor
interaction - Homo sapiens (human) | 0.047875420 | 1 | 1.319887 |
| Table VIPathway analysis of downregulated
mRNA in non-small cell lung cancer. |
Table VI
Pathway analysis of downregulated
mRNA in non-small cell lung cancer.
| Pathway ID | Definition | Fisher P-value | False discovery
rate | Enrichment
score |
|---|
| hsa05144 | Malaria - Homo
sapiens (human) | 7.58507E-06 | 0.001994873 | 5.120041 |
| hsa05143 | African
trypanosomiasis - Homo sapiens (human) | 0.000173854 | 0.022861780 | 3.759816 |
| hsa05330 | Allograft rejection
- Homo sapiens (human) | 0.000500645 | 0.043889850 | 3.300470 |
| hsa05310 | Asthma - Homo
sapiens (human) | 0.001029434 | 0.044896870 | 2.987401 |
| hsa05150 | Staphylococcus
aureus infection - Homo sapiens (human) | 0.001050144 | 0.044896870 | 2.978751 |
| hsa05416 | Viral myocarditis -
Homo sapiens (human) | 0.001179803 | 0.044896870 | 2.928191 |
| hsa04940 | Type I diabetes
mellitus - Homo sapiens (human) | 0.001194974 | 0.044896870 | 2.922642 |
| hsa05323 | Rheumatoid
arthritis - Homo sapiens (human) | 0.001701409 | 0.055933820 | 2.769191 |
| hsa05332 | Graft-versus-host
disease - Homo sapiens (human) | 0.004708912 | 0.137604900 | 2.327079 |
| hsa05140 | Leishmaniasis -
Homo sapiens (human) | 0.005457589 | 0.143534600 | 2.262999 |
| hsa00980 | Metabolism of
xenobiotics by cytochrome P450 - Homo sapiens (human) | 0.008647597 | 0.187985000 | 2.063105 |
| hsa04672 | Intestinal immune
network for IgA production - Homo sapiens (human) | 0.008855467 | 0.187985000 | 2.052789 |
| hsa05204 | Chemical
carcinogenesis - Homo sapiens (human) | 0.009292035 | 0.187985000 | 2.031889 |
| hsa04145 | Phagosome - Homo
sapiens (human) | 0.010739850 | 0.201755800 | 1.969002 |
| hsa04610 | Complement and
coagulation cascades - Homo sapiens (human) | 0.011933180 | 0.209228500 | 1.923244 |
| hsa05320 | Autoimmune thyroid
disease - Homo sapiens (human) | 0.013915240 | 0.215604300 | 1.856509 |
| hsa04640 | Hematopoietic cell
lineage - Homo sapiens (human) | 0.013936400 | 0.215604300 | 1.855849 |
| hsa04144 | Endocytosis -
Homo sapiens (human) | 0.015738420 | 0.220986000 | 1.803039 |
| hsa00982 | Drug
metabolism-cytochrome P450 - Homo sapiens (human) | 0.015964770 | 0.220986000 | 1.796837 |
| hsa04062 | Chemokine signaling
pathway - Homo sapiens (human) | 0.018525790 | 0.234434600 | 1.732223 |
| hsa04060 | Cytokine-cytokine
receptor interaction - Homo sapiens (human) | 0.018719110 | 0.234434600 | 1.727715 |
| hsa00071 | Fatty acid
metabolism - Homo sapiens (human) | 0.020507740 | 0.245160700 | 1.688082 |
| hsa05152 | Tuberculosis -
Homo sapiens (human) | 0.032055540 | 0.366548100 | 1.494097 |
| hsa05142 | Chagas disease
(American trypanosomiasis) - Homo sapiens (human) | 0.034257470 | 0.375404800 | 1.465245 |
| hsa04514 | Cell adhesion
molecules (CAMs) - Homo sapiens (human) | 0.037073840 | 0.389678400 | 1.430932 |
| hsa00900 | Terpenoid backbone
biosynthesis - Homo sapiens (human) | 0.038523340 | 0.389678400 | 1.414276 |
| hsa03320 | PPAR signaling
pathway - Homo sapiens (human) | 0.042811800 | 0.412312800 | 1.368437 |
| hsa04970 | Salivary secretion
- Homo sapiens (human) | 0.043896420 | 0.412312800 | 1.357571 |
RT-qPCR validation
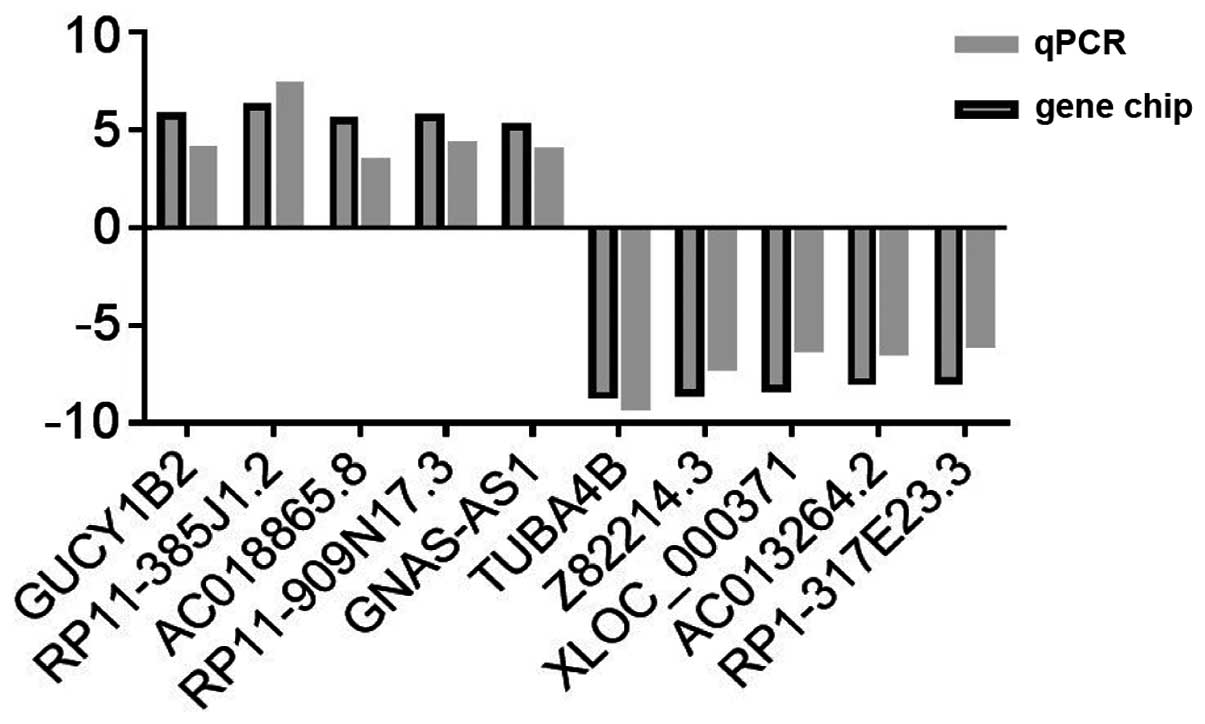
According to factors including the fold difference,
gene locus and nearby encoding gene, we initially identified a
number of significant candidate lncRNAs (including GUCY1B2,
RP11-385J1.2, AC018865.8, RP11-909N17.3, GNAS-AS1, TUBA4B,
Z82214.3, XLOC_000371, AC013264.2 and RP1-317E23.3) and verified
the expression of these lncRNAs by RT-qPCR with GAPDH as the
reference gene, by calculating the 2−ΔΔCT values. We
observed that multiple lncRNAs in the microarray were consistent
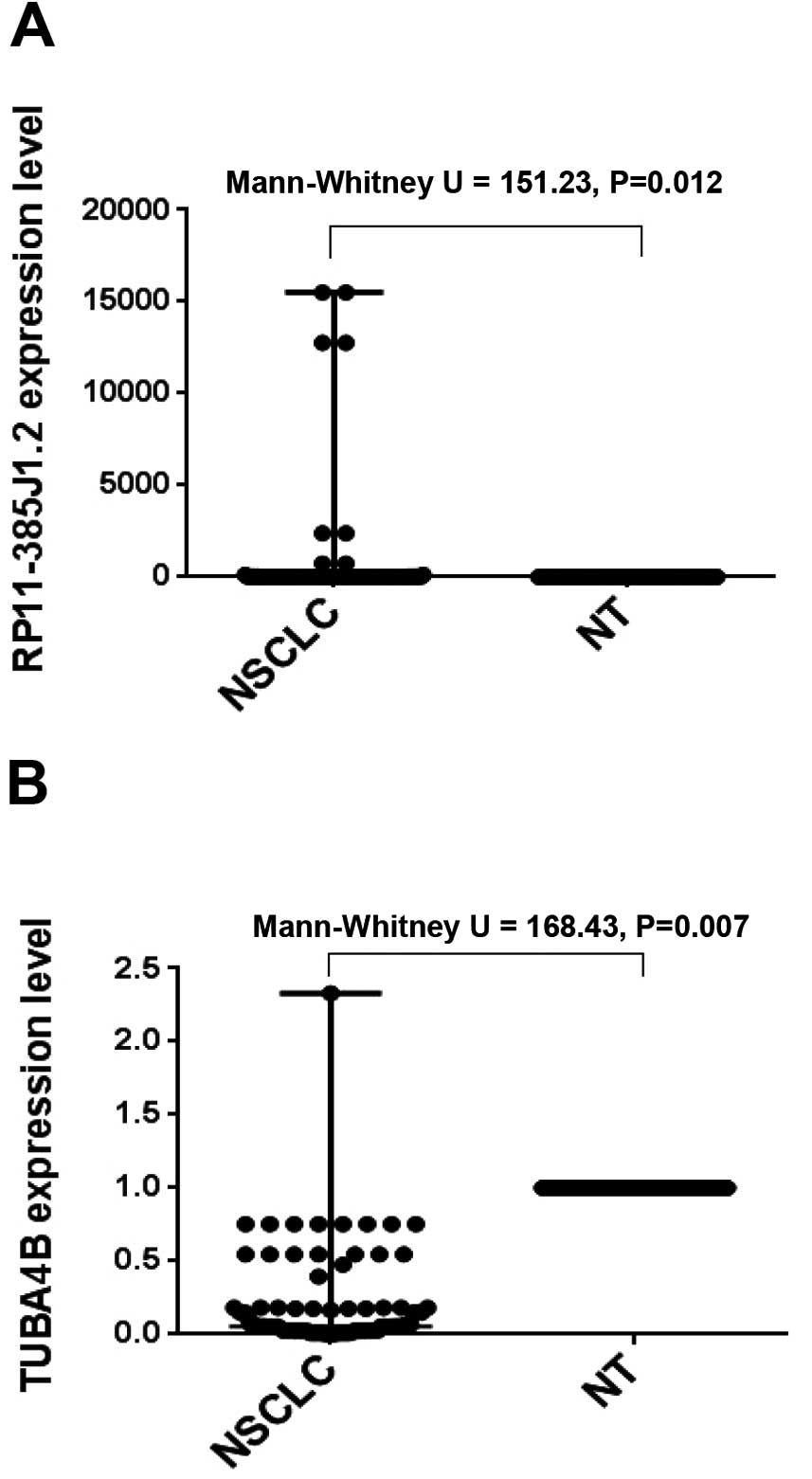
with the results of the RT-qPCR; see Fig. 3. RP11-385J1.2 and TUBA4B were the
most markedly changed of these candidate lncRNAs from 90 NSCLC and
normal lung tissue samples. As shown in Fig. 4, RP11-385J1.2 expression in NSCLC
was significantly higher than in the adjacent tissues (Mann-Whitney
U test=151.23, P=0.012), while TUBA4B expression in NSCLC was
significantly lower than in the adjacent tissues (Mann-Whitney U
test=168.43, P=0.007).
Discussion
According to the 2012 China Oncology Annual Report,
the 2009 incidence and mortality of lung cancer was the highest
among all cancers in male patients and the second highest among all
cancers in female patients in China. LncRNAs play a significant
role in a number of biological processes, including X-chromosome
inactivation, gene imprinting and stem cell maintenance (24,25).
It has been confirmed that lncRNAs are one of the most significant
factors controlling gene expression in cancer (26). LncRNAs including HOTAIR have been
shown to play a crucial role in the development and progression of
tumors (9). It has also been
demonstrated that lncRNAs are differentially expressed in normal
and tumor cells (27,28). As lncRNAs constitute an essential
class of gene expression regulatory factors, their aberrant
expression would inevitably lead to abnormal gene expression
levels, which may result in tumorigenesis.
In this study, we analyzed the lncRNA expression
profile in the tissue of NSCLC patients to elucidate the potential
role of lncRNAs in the pathogenesis of this disease.
High-throughput microarray techniques revealed a set of
differentially expressed lncRNAs, with 541 of those upregulated and
701 downregulated in NSCLC tissue compared with normal lung tissue.
LncRNAs are usually divided into five categories: Sense, antisense,
bidirectional, intronic and intergenic (29). LncRNAs are known to function via a
variety of mechanisms; however, a common and significant function
of lncRNAs is to alter the expression of nearby encoding genes by
affecting the process of transcription (30) or directly playing an enhancer-like
role (31,32). In the present study, we increased
the accuracy of target prediction by comparing differentially
expressed mRNAs with differentially expressed lncRNAs. The lncRNA
expression profiles indicated that 343 lncRNAs were differentially
expressed (167 upregulated and 176 downregulated) between NSCLC and
normal lung samples. The expression profiles included 18
differentially expressed enhancer-like lncRNAs, with seven
upregulated and 11 downregulated. Nearby coding genes that may be
regulated by lncRNAs and enhancer-like lncRNAs were also
identified. In addition, we performed HOX cluster profiling of
lncRNAs and coding transcripts.
In order to obtain insights into lncRNA target gene
function, GO analysis and KEGG pathway annotation were applied to
the lncRNA target gene pool. GO analysis revealed that the number
of genes corresponding to downregulated mRNAs was larger than that
corresponding to upregulated mRNAs. KEGG annotation revealed 11
upregulated pathways (including ethanol metabolism, systemic lupus
erythematosus, transcriptional misregulation in cancer and cell
cycle pathways) and 28 downregulated pathways (including malaria,
African trypanosomiasis and allograft rejection). These pathways
may play significant roles in the occurrence and development of
NSCLC. Ten lncRNAs identified in the microarray analysis were
confirmed by RT-qPCR to be aberrantly expressed in NSCLC tissues.
Among these lncRNAs, RP11-385J1.2 was the most markedly upregulated
and TUBA4B was the most markedly downregulated. This result
suggests that RP11-385J1.2 and TUBA4B may contribute to the
development of NSCLC; further study of the biological function of
RP11-385J1.2 and TUBA4B will be required to confirm this.
In conclusion, the present study revealed a set of
lncRNAs with differential expression in NSCLC compared with normal
lung tissue. Furthermore, it was demonstrated that RP11-385J1.2 and
TUBA4B may contribute to the development of NSCLC. Further
investigation of the lncRNAs identified in this study will likely
provide insights into their biological functions and their
association with NSCLC.
Acknowledgements
This study was financially supported by the National
Natural Science Foundation of China (81401736 and 81271906) and
Wenzhou Municipal Science and Technology Bureau, China (Y20110041
and Y20130170). The authors thank all donors who donated to the
microarray service at KangChen Bio-tech, Shanghai, China.
References
|
1
|
Jemal A, Murray T, Ward E, Samuels A,
Tiwari RC, Ghafoor A, Feuer EJ and Thun MJ: Cancer statistics,
2005. CA Cancer J Clin. 55:10–30. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gridelli C, Rossi A and Maione P:
Treatment of non-small-cell lung cancer: state of the art and
development of new biologic agents. Oncogene. 22:6629–6638. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stewart DJ: Tumor and host factors that
may limit efficacy of chemotherapy in non-small cell and small cell
lung cancer. Crit Rev Oncol Hematol. 75:173–234. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen CH, Lai JM, Chou TY, Chen CY, Su LJ,
Lee YC, et al: VEGFA upregulates FLJ10540 and modulates migration
and invasion of lung cancer via PI3K/AKT pathway. PloS One.
4:e50522009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ogawa E, Takenaka K, Katakura H, Adachi M,
Otake Y, Toda Y, et al: Perimembrane Aurora-A expression is a
significant prognostic factor in correlation with proliferative
activity in non-small-cell lung cancer (NSCLC). Ann Surg Oncol.
15:547–554. 2008. View Article : Google Scholar :
|
|
6
|
Rachet B, Woods LM, Mitry E, Riga M,
Cooper N, Quinn MJ, Coleman MP, et al: Cancer survival in England
and Wales at the end of the 20th century. Br J Cancer. 99:S2–S10.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ponting CP, Oliver PL and Reik W:
Evolution and functions of long noncoding RNAs. Cell. 136:629–641.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dinger ME, Pang KC, Mercer TR, Crowe ML,
Grimmond SM and Mattick JS: NRED: a database of long noncoding RNA
expression. Nucleic Acids Res. 37:D122–D126. 2009. View Article : Google Scholar :
|
|
9
|
Gupta RA, Shah N, Wang KC, Kim J, Horlings
HM, Wong DJ, Chang HY, et al: Long non-coding RNA HOTAIR reprograms
chromatin state to promote cancer metastasis. Nature.
464:1071–1076. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wapinski O and Chang HY: Long noncoding
RNAs and human disease. Trends Cell Biol. 21:354–361. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fu X, Ravindranath L, Tran N, Petrovics G
and Srivastava S: Regulation of apoptosis by a prostate-specific
and prostate cancer-associated noncoding gene, PCGEM1. DNA Cell
Biol. 25:135–141. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang H, Chen Z, Wang X, Huang Z, He Z and
Chen Y: Long non-coding RNA: a new player in cancer. J Hematol
Oncol. 6:372013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hauptman N and Glavac D: Long non-coding
RNA in cancer. Int J Mol Sci. 14:4655–4669. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen G, Wang Z, Wang D, Qiu C, Liu M, Chen
X, Cui Q, et al: LncRNADisease: a database for long-non-coding
RNA-associated diseases. Nucleic Acids Res. 41:D983–D986. 2013.
View Article : Google Scholar :
|
|
15
|
Gibb EA, Brown CJ and Lam WL: The
functional role of long non-coding RNA in human carcinomas. Mol
Cancer. 10:382011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ji P, Diederichs S, Wang W, Boing S,
Metzger R, Schneider PM, Muller-Tidow C, et al: MALAT-1, a novel
noncoding RNA, and thymosin beta4 predict metastasis and survival
in early-stage non-small cell lung cancer. Oncogene. 22:8031–8041.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Thai P, Statt S, Chen CH, Liang E,
Campbell C and Wu R: Characterization of a novel long noncoding
RNA, SCAL1, induced by cigarette smoke and elevated in lung cancer
cell lines. Am J Respir Cell Mol Biol. 49:204–211. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang Y, Li H, Hou S, Hu B, Liu J and Wang
J: The noncoding RNA expression profile and the effect of lncRNA
AK126698 on cisplatin resistance in non-small-cell lung cancer
cell. PloS One. 8:e653092013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Han L, Kong R, Yin DD, Zhang EB, Xu TP, De
W and Shu YQ: Low expression of long noncoding RNA GAS6-AS1
predicts a poor prognosis in patients with NSCLC. Med Oncol.
30:6942013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fritah S, Niclou SP and Azuaje F:
Databases for lncRNAs: a comparative evaluation of emerging tools.
RNA. 20:1655–1665. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chakraborty S, Deb A, Maji RK, Saha S and
Ghosh Z: LncRBase: an enriched resource for lncRNA information.
PLoS One. 9:e1080102014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Quek XC, Thomson DW, Maag JL, et al:
lncRNAdb v2.0: expanding the reference database for functional long
noncoding RNAs. Nucleic Acids Res. Oct 20–2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ren S, Peng Z, Mao JH, Yu Y, Yin C, Gao X,
Sun Y, et al: RNA-seq analysis of prostate cancer in the Chinese
population identifies recurrent gene fusions, cancer-associated
long noncoding RNAs and aberrant alternative splicings. Cell Res.
22:806–821. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mercer TR, Dinger ME and Mattick JS: Long
non-coding RNAs: insights into functions. Nature reviews Genetics.
10:155–159. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang KC and Chang HY: Molecular mechanisms
of long noncoding RNAs. Mol Cell. 43:904–914. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Khachane AN and Harrison PM: Mining
mammalian transcript data for functional long non-coding RNAs. PloS
One. 5:e103162010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lai MC, Yang Z, Zhou L, et al: Long
non-coding RNA MALAT-1 overexpression predicts tumor recurrence of
hepatocellular carcinoma after liver transplantation. Med Oncol.
29:1810–1816. 2012. View Article : Google Scholar
|
|
28
|
Braconi C, Kogure T, Valeri N, et al:
microRNA-29 can regulate expression of the long non-coding RNA gene
MEG3 in hepatocellular cancer. Oncogene. 30:4750–4756. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li CH and Chen Y: Targeting long
non-coding RNAs in cancers: progress and prospects. Int J Biochem
Cell Biol. 45:1895–1910. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mattick JS and Gagen MJ: The evolution of
controlled multitasked gene networks: the role of introns and other
noncoding RNAs in the development of complex organisms. Mol Biol
Evol. 18:1611–1630. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mattick JS: Linc-ing Long noncoding RNAs
and enhancer function. Dev Cell. 19:485–486. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Orom UA, Derrien T, Beringer M, Gumireddy
K, Gardini A, Bussotti G, Shiekhattar R, et al: Long noncoding RNAs
with enhancer-like function in human cells. Cell. 143:46–58. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Edge SB and Compton CC: The American Joint
Committee on Cancer: the 7th edition of the AJCC cancer staging
manual and the future of TNM. Ann Surg Oncol. 17:1471–1474. 2010.
View Article : Google Scholar : PubMed/NCBI
|