Introduction
Changes in the extracellular matrix (ECM) have been
associated with numerous pathologies, including atherosclerosis
(AS). Increased deposition of ECM follows various phases, including
formation of fatty streaks and fibrous plaques, which can result in
AS. A reduction in the ECM can result in the erosion and cracking
of fibrous plaques, leading to myocardial infarction. The balance
between the enzymes functioning to degrade the ECM and endogenous
inhibitors is an important factor in determining the progression of
AS and plaque stability (1). MMPs
are the predominant enzymes which function to degrade the vascular
ECM. The activation and proteolytic function of MMPs is regulated
by tissue inhibitors of metalloproteinase-1 (TIMPs). Previous
studies have suggested that MMP-9 and associated endogenous
inhibitor TIMP-1 function across all stages of AS progression
(1). Smooth muscle cells are the
only cellular components of the arterial wall membrane of mammals.
It has been previously confirmed that vascular smooth muscle cells
(VSMCs) migrate from the arterial tunica media to the tunica
intima, which results in phenotypic changes to the arterial wall
membrane, and abundant proliferation and the formation of myogenic
foam cells. This is important in the pathological development of AS
(2,3). Newly derived VSMCs only have the
capacity for binary fission, but secrete large amounts of ECM and
active substances, including MMPs and TIMPs (4,5).
Studying the impact of the various risk factors that promote the
secretion of MMPs and TIMPs by VSMCs may be useful in the
understanding of the pathogenesis of coronary heart disease. It is
currently considered that the renin-angiotensin (Ang)-aldosterone
system (RAAS) is involved in the pathological process of AS, in
which AngII has a central part. Previous studies have suggested
that losartan, an AngII receptor (AT1) antagonist, produces
anti-arteriosclerosis effects. The present study therefore
hypothesized that AngII and losartan may affect the secretion of
MMPs and TIMPs by VSMCs, thus functioning in anti-AS or
AS-induction (6–8). In the present study, rat VSMCs were
cultured in vitro and analyzed for the effects of losartan
and AngII in the secretion of MMP-9 and TIMP-1. The present study
aimed to demonstrate the AS-induction effect of AngII and anti-AS
effect of losartan.
Materials and methods
Primary cultivation of the adherent
tissue blocks
The study was approved by the ethics committee of
the Second Military Medical University (Shanghai, China). Male
Wistar rats were obtained from the Animal Center of Shanghai Second
Medical Military University (weight, 200–300 g; age, three-four
months). They were fed with a standard diet and water, and housed
at a temperature of 21–27°C. The thoracic aorta was surgically
isolated from a healthy male Wistar rat. The adventitia and intima
was removed, and the media layer was cut in tissue blocks sized ~1
mm3. The tissue blocks were transferred onto the walls
of a 25 cm2 plastic culture flask, to which 5 ml
Dulbecco’s modified Eagle’s medium (DMEM) with 20% newborn calf
serum (NCS; Hangzhou Evergreen Company, Hangzhou, China)
inactivated at 56°C for 0.5 h followed by packaging and
preservation at 21°C (Hangzhou Evergreen Company), was added to the
contralateral bottom. The flask was sealed and incubated at 37°C
with 5% CO2 for 4 h. Following this, the culture flask
was gently flipped for static cell culture. Following one week of
culture, VSMCs were observed growing from the tissues, and after
2–3 weeks, a fused dense monolayer of proliferating cells formed.
The cells were digested with 0.1% trypsin for passaging. The fourth
to 10th generations of smooth muscle cells (SMCs) were obtained for
subsequent experiments or frozen in liquid nitrogen.
Cell synchronization
Following 3–4 days of subculture, synchronization
was performed according to the requirements of the experiment. The
supernatant was decanted, and the cells were washed with
phosphate-buffered saline (PBS) 2–3 times. The cells were then
added to the DMEM containing 0.5% NBS, which restrained the
majority of cells to the G0 phase. When required, DMEM
containing 20% NBS could be used to force the cells to proliferate
(DNA synthesis phase).
Identification of VSMCs
An inverted phase contrast microscope (CKX31-A12PHP;
Olympus Corporation, Tokyo, Japan) was used to observe the
morphology and growth patterns of living cells.
Immunohistochemistry staining was used for detection of
anti-α-actin as a specific indicator for VSMCs. Under sterile
conditions, cover slips were used to cover the 6-well cell culture
plates for VSMCs seeding. Following 48 hr of cultivation, the cover
slips were removed and samples were washed three times for one min
with PBS buffer, followed by fixation with 95% alcohol for 20 min.
The streptavidin-peroxidase immunohistochemical method was then
performed. This involved washing three times with PBS buffer, then
soaking in 3% peroxidized methanol at room temperature for 20 min.
Samples were then washed again three times with PBS buffer prior to
addition of 50 μl 5% normal goat serum, followed by incubation at
room temperature for 10 min. Mouse anti-rat SMA monoclonal antibody
(50 μl, 1:500; Sigma) was added dropwise, and samples were then
cultured at room temperature for 60 minutes. Following three washes
with PBS, 50 μl biotinylated monoclonal goat anti-mouse
immunoglobulin (Ig)G secondary antibody (1:200; Sigma) was added
dropwise and samples were cultured at room temperature for 10 min.
Sample were then washed three times with PBS, 50 μl streptomycin
avidin-peroxidase solution was added followed by culturing at room
temperature for 10 min. Freshly-configured DAB solution (100 μl)
was added dropwise. Samples were then washed with water, restained
with hematoxylin and mounted on neutral gum prior to observation
under the microscope.
Western blot analysis
The isolated cultured cells were seeded in culture
plates at a concentration of 1×106/ml. Following
synchronization, the medium was replaced with DMEM containing 2%
newborn calf serum (NCS). Different concentrations of losartan
(Merck Co., Ltd., Whitehouse Station, NJ, USA) were added to the
wells to produce final concentrations of 10−7,
10−6, 10−5 and 10−4 M. A well
without addition of losartan was considered to be a blank control.
The culture plate was incubated at 37°C and 5% CO2 for
24 h. The cells were then cultivated by collecting the supernatant
and using a vacuum dryer to concentrate the samples. Coomassie blue
staining method was used to determine the protein concentration. A
protein sample of 20 μg was used for SDS-PAGE and western blotting.
The procedure for western blotting was as follows. Thoracic aorta
(50 mg)was cut into small pieces using a scalpel. PBS (1 ml) was
added and tissues were homogenized twenty times using a glass
homogenizer in an ice bath, followed by centrifugation at 2,124 × g
at 4°C for 10 min. The supernatant was then collected and stored at
−80°C. SDS-PAGE gel electrophoresis was used to separate the
proteins. Samples were transferred on to a cellulose nitrate film
(Hybond™-C; Amersham Biosciences, Piscataway, NJ, USA). Following
transfer, the nitrocellulose membrane was placed into the closure
solution (double distilled water) for 2 h. The closure solution was
discarded and the membrane was washed three times with 0.1% (v/v)
Tween-20 in Tris-buffered saline (TTBS; Biosharp, St. Louis, MO,
USA) solution. TTBS was discarded and the primary mouse anti-rat
MMP-9/TIMP-1 monoclonal antibody (diluted 1:500 with TBS) was added
and incubated for 2 h. The primary antibody was then discarded and
the membrane was rinsed three times with TTBS. TTBS was then
discarded and the secondary alkaline phosphatase conjugated goat
anti-mouse IgG monoclonal antibody (diluted 1:500 in TBS) was added
and incubated for 2 h. The secondary antibody was then discarded,
and the membrane was rinsed twice with TTBS and once with PBS. The
appropriate amount of the staining reagents ) nitro-blue
tetrazolium (NBT) and bromo chloro indole phosphate (BCIP),
obtained from Sigma, were added, according to the manufacturer’s
instructions and the colored bands were observed within 20 min.
Distilled water was used to stop the reaction. The resultant bands
were visualized with radiography and pictures were captured,
scanned and analyzed using a computer image analyzer gel imaging
system (GDS8000; Ultra-Violet Products, Upland, CA, USA). According
to the instructions of the GDS8000 gel imaging system, the gray
values of each signal band were then determined. Experiments were
repeated four times, and the value of the control group was set as
1 for the calculation of each group’s relative value.
Quantitative polymerase chain reaction
(qPCR) and agarose gel electrophoresis
qPCR primers were synthesized by Shanghai Boya
Biotechnology Co., Ltd. (Shanghai, China). The sequences were as
follows: MMP-9 sense, 5′-GGCCTATTTCTGCCATGACAAATAC-3′ and
antisense, 5′-CTGCACCGCTGAAGCAAAAG-3′; TIMP-1 sense,
5′-CCCCAGAAATCATCGAGAC-3′ and antisense, 5′-GATTATGCCAGGGAACCAG-3′;
β-actin sense, 5′-GTGGGGCGCCCCAGGCACCA-3′ and antisense,
5′-CTCCTTAATTGTCACGCACGATTC-3′. Total RNA was extracted, followed
using one-step RT-PCR kits obtained from Roche Diagnostics
(Mannheim, Germany), according to the manufacturer’s instructions.
The following conditions were used: 50°C for 60 min; 94°C for 30
min; 30 cycles of 94° for 30 sec, then 55°C for 30 sec, then 72°
for 1 min; and 72°C for 7 min. A 10-μl sample of qPCR product and
β-actin of the same sample were added to 4 μl of 6X bromophenol
blue buffer. The samples were then separated by 1% agarose gel
(0.5X TAE) electrophoresis at 100 V. After 1 h, nucleotides stained
with bromophenol blue had migrated 3/4 of the total distance in the
gel, and the electrophoresis was ended. The gels were imaged and a
computer image analyzer scanned and analyzed the obtained bands. A
comparison of the grayscale signals of each band were madde using a
gel image analyzer (GDS8000) in order to semi-quantitively analyze
the samples. This was repeated four times. The value of the control
group was set as 1 for the subsequent calculation of the value of
each group.
Statistical analysis
The data are expressed as the mean ± standard
deviation. An analysis of variance was performed where appropriate,
and the Student–Newman–Keuls method was used for pairwise
comparison. P<0.05 was considered to indicate a statistically
significant difference.
Results
Identification of VSMCs
The SMC’s typical growth pattern was observed once
the cells had grown and covered the bottom of the flask. The cells
appeared spindle-shaped, growing in a parallel fashion, and
arranged in cellular bunches. The dense and sparse areas overlapped
and appeared in a “peak-valley” conformation.
Immunohistochemical staining used an antibody
against smooth muscle actin (SMA; Sigma, St Louis, MO, USA). The
cells were stained brown, giving positive confirmation that the
cultured cells were VSMCs.
Western blot analysis
The effects of different AngII concentrations on the
expression of MMP-9 and TIMP-1 protein in rat aortic VSMCs are
shown in Fig. 1A. AngII could
promote the expression of MMP-9, thus increasing the MMP-9/TIMP-1
ratio, in a concentration-dependent manner. AngII had no effect on
the expression of TIMP-1 (Table
I).
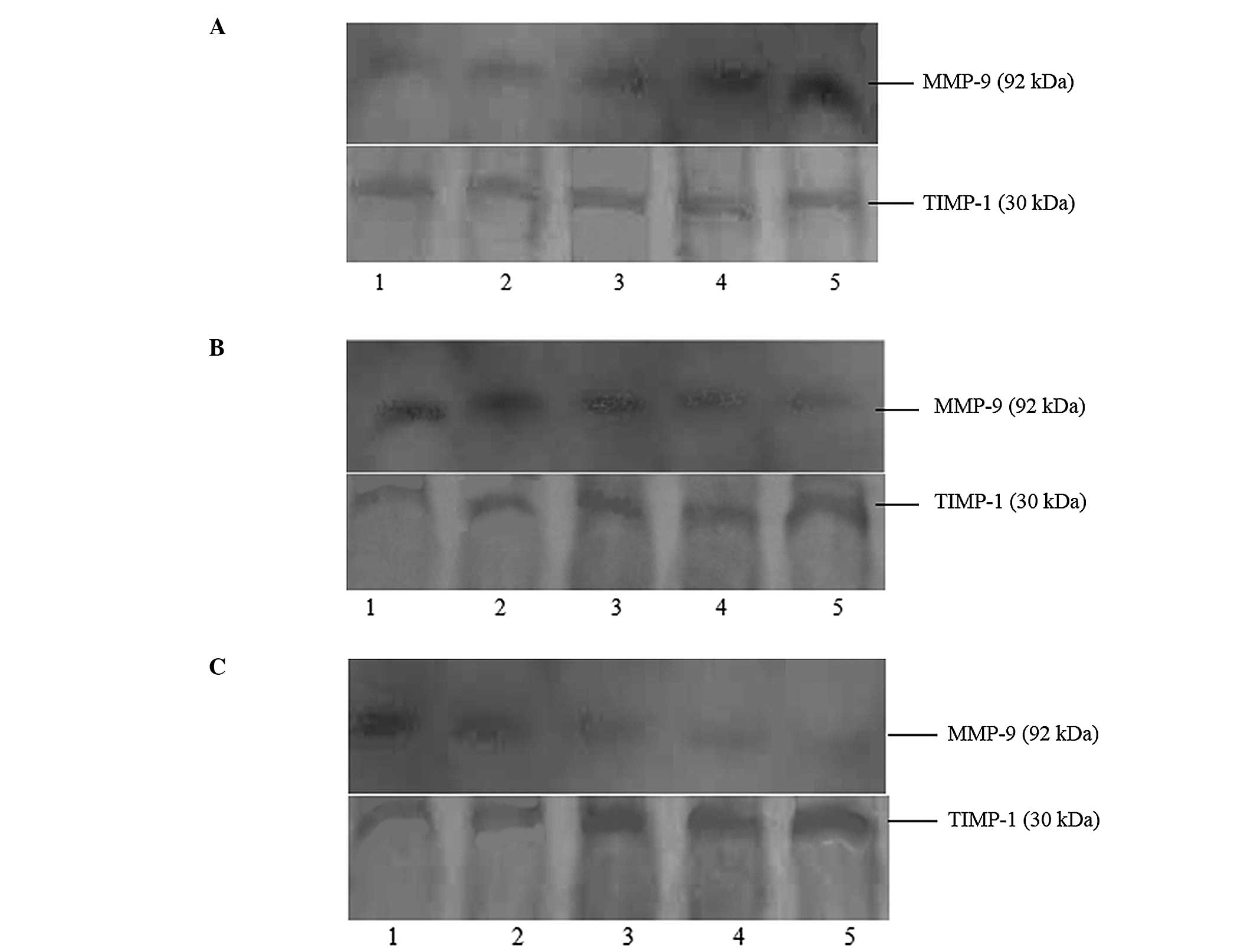 | Figure 1Effects of Ang II/losartan/losartan +
AngII with different concentrations on expressions of MMP-9 and
TIMP-1 protein in rat aortic vascular smooth muscle cells (24-h
incubation). (A) Lanes 1–5 presented as blank, AngII
10−9 M, AngII 10−8 M, AngII 10−7 M
and AngII 10−6 M, respectively. (B) Lanes 1–5 presented
as blank, losartan 10−7 M, losartan 10−6 M,
losartan 10−5 M and losartan 10−4 M,
respectively. (C) Lane 1 presented as blank, and lanes 2–5
presented as added losartan 10−7 M, losartan
10−6 M, losartan 10−5 M and losartan
10−4 M based on AngII 10−6 M, respectively.
MMP-9, matrix metalloproteinase-9; TIMP-1, tissue inhibitor of
metalloproteinase-1; AngII, angiotensin II. |
 | Table IComparison of the gradation values of
the obtained protein bands (Fig.
1A) as a result of different angiotensin II concentrations
towards the expression of MMP-9 and TIMP-1 proteins in rat aortic
vascular smooth muscle cells. |
Table I
Comparison of the gradation values of
the obtained protein bands (Fig.
1A) as a result of different angiotensin II concentrations
towards the expression of MMP-9 and TIMP-1 proteins in rat aortic
vascular smooth muscle cells.
| Groups | Control | 10−9
M | 10−8
M | 10−7
M | 10−6
M |
|---|
| MMP-9 gradation | 1 | 1.06±0.08 | 1.35±0.11a | 1.69±0.13a | 2.22±0.18a |
| TIMP-1 gradation | 1 | 0.98±0.08 | 1.05±0.11 | 0.98±0.10 | 1.04±0.12 |
| MMP-9/TIMP-1 | 1 | 1.05±0.07 | 1.29±0.11a | 1.72±0.13a | 2.13±0.18a |
The effects of different concentrations of losartan
on the expression of MMP-9 and TIMP-1 protein in rat aortic VSMCs
are shown in Fig. 1B. Losartan
inhibited the expression of MMP-9 and promoted the expression of
TIMP-1. This resulted in a decrease in the MMP-9/TIMP-1 ratio in a
concentration-dependent manner (Table
II).
| Table IIComparison of the gradation values of
the obtained protein bands (Fig.
1B) as a result of different losartan concentrations towards
the expressions of MMP-9 and TIMP-1 proteins in rat aortic vascular
smooth muscle cells. |
Table II
Comparison of the gradation values of
the obtained protein bands (Fig.
1B) as a result of different losartan concentrations towards
the expressions of MMP-9 and TIMP-1 proteins in rat aortic vascular
smooth muscle cells.
| Groups | Control | 10−7
M | 10−6
M | 10−5
M | 10−4
M |
|---|
| MMP-9 gradation | 1 | 0.90±0.10 | 0.72±0.11a | 0.58±0.13a | 0.38±0.15a |
| TIMP-1 gradation | 1 | 1.26±0.13a | 1.63±0.13a | 1.90±0.13a | 2.19±0.13a |
| MMP-9/TIMP-1 | 1 | 0.71±0.06a | 0.44±0.07a | 0.31±0.05a | 0.17±0.07a |
The effects of the combination of losartan and AngII
on the expression of MMP-9 and TIMP-1 protein in rat aortic VSMCs
are shown in Fig. 1C. The
combination of AngII and losartan inhibited the expression of MMP-9
and promoted the expression of TIMP-1, therefore decreasing the
MMP-9/TIMP-1 ratio, in a losartan-concentration-dependent manner
(Table III).
| Table IIIComparison of the gradation values of
the obtained protein bands (Fig.
1C) as a result of the combination of different losartan
concentrations and angiotensin II towards the expressions of MMP-9
and TIMP-1 proteins in rat aortic vascular smooth muscle cells. |
Table III
Comparison of the gradation values of
the obtained protein bands (Fig.
1C) as a result of the combination of different losartan
concentrations and angiotensin II towards the expressions of MMP-9
and TIMP-1 proteins in rat aortic vascular smooth muscle cells.
| Groups | Control | 10−7
M | 10−6
M | 10−5
M | 10−4
M |
|---|
| MMP-9
gradation | 1 | 0.93±0.07 | 0.7±0.06a | 0.59±0.05a | 0.37±0.04a |
| TIMP-1
gradation | 1 | 1.28±0.09a | 1.61±0.14a | 1.88±0.17a | 2.01±0.19a |
| MMP-9/TIMP-1 | 1 | 0.73±0.06a | 0.43±0.04a | 0.31±0.04a | 0.18±0.02a |
qPCR analysis
The effects of different AngII concentrations on the
expression of MMP-9 and TIMP-1 mRNA in rat aortic VSMCs are shown
in Fig. 2A and B. AngII could
promote the expression of MMP-9 mRNA, increasing the MMP-9/TIMP-1
ratio, in a concentration-dependent manner. No effect of AngII was
observed on the expression of TIMP-1 mRNA (Table IV).
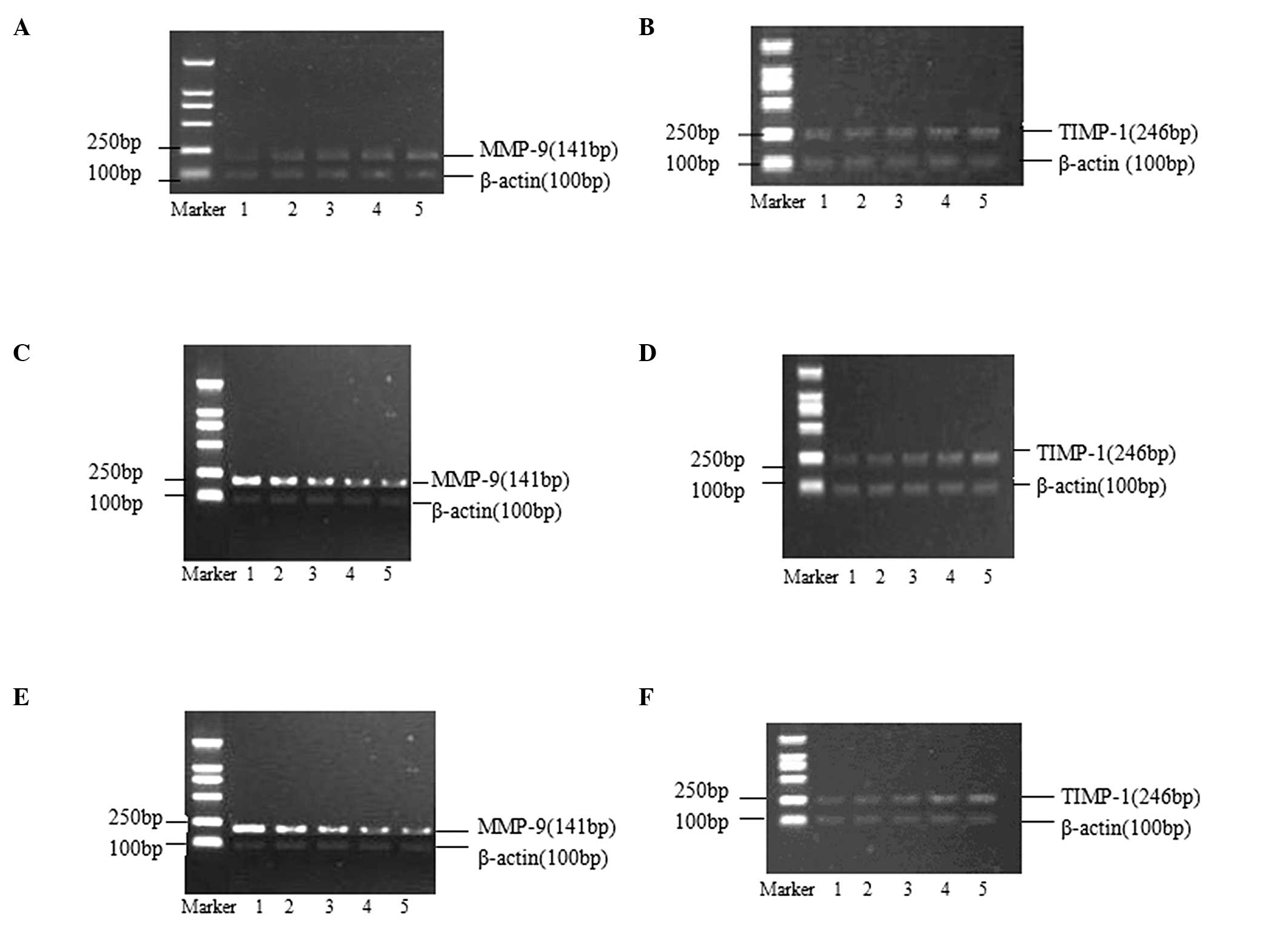 | Figure 2Effects of AngII/losartan/losartan +
AngII with different concentrations on expressions of MMP-9 and
TIMP-1 mRNA in rat aortic vascular smooth muscle cells (24-h
incubation). (A and B) Lanes 1–5 presented as blank, AngII
10−9 M, AngII 10−8 M, AngII 10−7 M
and AngII 10−6 M, respectively. (C and D) Lanes 1–5
presented as blank, losartan 10−7 M, losartan
10−6 M, losartan 10−5 M and losartan
10−4 M, respectively. (E and F) Lane 1 presented as
blank, and lanes 2–5 presented added losartan 10−7 M,
losartan 10−6 M, losartan 10−5 M and losartan
10−4 M based on AngII 10−6 M, respectively.
MMP-9, matrix metalloproteinase-9; TIMP-1, tissue inhibitor of
metalloproteinase-1. AngII, angiotensin II. |
| Table IVComparison of the gradation values of
the obtained bands (Fig. 2A and B)
as a result of different angiotensin II concentrations towards the
expressions of MMP-9 mRNA and TIMP-1 mRNA in rat aortic vascular
smooth muscle cells. |
Table IV
Comparison of the gradation values of
the obtained bands (Fig. 2A and B)
as a result of different angiotensin II concentrations towards the
expressions of MMP-9 mRNA and TIMP-1 mRNA in rat aortic vascular
smooth muscle cells.
| Groups | Control | 10−9
M | 10−8
M | 10−7
M | 10−6
M |
|---|
| MMP-9
gradation | 1 | 1.28±0.11a | 1.36±0.11a | 1.58±0.10a | 1.92±0.13a |
| TIMP-1
gradation | 1 | 1.01±0.08 | 1.04±0.11 | 0.98±0.10 | 0.94±0.18 |
| MMP-9/TIMP-1 | 1 | 1.27±0.04a | 1.31±0.05a | 1.61±0.06a | 2.04±0.04a |
The effects of different concentrations of losratan
on the expression of MMP-9 and TIMP-1 mRNA in rat aortic VSMCs are
shown in Fig. 2C and D. Losartan
inhibited the expression of MMP-9 mRNA and promoted the expression
of TIMP-1 mRNA, therefore decreasing the MMP-9/TIMP-1 ratio, in a
concentration-dependent manner (Table
V).
| Table VComparison of the gradation values of
the obtained bands (Fig. 2C and D)
as a result of different losartan concentrations towards the
expressions of MMP-9 mRNA and TIMP-1 mRNA in rat aortic vascular
smooth muscle cells. |
Table V
Comparison of the gradation values of
the obtained bands (Fig. 2C and D)
as a result of different losartan concentrations towards the
expressions of MMP-9 mRNA and TIMP-1 mRNA in rat aortic vascular
smooth muscle cells.
| Groups | Control | 10−7
M | 10−6
M | 10−5
M | 10−4
M |
|---|
| MMP-9
gradation | 1 | 0.91±0.10 | 0.69±0.11a | 0.47±0.13a | 0.31±0.15a |
| TIMP-1
gradation | 1 | 1.26±0.13a | 1.53±0.13a | 1.80±0.13a | 2.03±0.13a |
| MMP-9/TIMP-1 | 1 | 0.72±0.06a | 0.45±0.07a | 0.26±0.05a | 0.15±0.07a |
The effects of the combination of losartan and AngII
on the expression of MMP-9 and TIMP-1 mRNA in rat aortic VSMCs are
shown in Fig. 2E and F. The
combination was observed to inhibit the expression of MMP-9 mRNA
and to promote the expression of TIMP-1 mRNA, therefore decreasing
the MMP-9/TIMP-1 ratio, in a losartan-concentration-dependent
manner (Table VI).
| Table VIComparison of the gradation values of
the obtained bands (Fig. 2E and F)
as a result of the combination of different losartan concentrations
and angiotensin II towards the expressions of MMP-9 mRNA and TIMP-1
mRNA in rat aortic vascular smooth muscle cells. |
Table VI
Comparison of the gradation values of
the obtained bands (Fig. 2E and F)
as a result of the combination of different losartan concentrations
and angiotensin II towards the expressions of MMP-9 mRNA and TIMP-1
mRNA in rat aortic vascular smooth muscle cells.
| Groups | Control | 10−7
M | 10−6
M | 10−5
M | 10−4
M |
|---|
| MMP-9
gradation | 1 | 0.91±0.13 | 0.74±0.11a | 0.59±0.10a | 0.41±0.12a |
| TIMP-1
gradation | 1 | 1.07±0.09 | 1.27±0.10a | 1.77±0.12a | 1.91±0.13a |
| MMP-9/TIMP-1 | 1 | 0.85±0.04 | 0.81±0.05b | 0.76±0.06a | 0.70±0.05a |
Discussion
The formation of AS is a slow process that may
require decades in order to develop from initial fatty streaks to
the advanced complex plaques. The progression of AS lesions is a
consequence of three predominant processes. Firstly, damage to the
arterial intima may cause lipid infiltration and deposition under
the intima, accumulating cholesterol ester and free cholesterol in
the cells and the surrounding connective tissue matrices. Secondly,
a large number of VSMCs, macrophages and T lymphocytes may
accumulate in the intima. Thirdly, VSMCs can migrate from the media
to the intima, where they undergo large-scale proliferation,
generating a large quantity of ECM and active substances. These
biological processes are all closely associated with changes to the
ECM. The ECM is an insoluble structural component that comprises
the interstitial tissue and vascular matrix. The ECM has a physical
and mechanical function in support of tissues and cells, as well as
functioning to regulate the healing and fibration of tissue wounds,
aging and cancer process of cells (9,10).
The balance of the enzymes degrading the ECM and endogenous
inhibitors are important in determining the progression of AS and
plaque stability. During the development of AS, VSMC proliferation
occurs across various periods of AS lesions, in the stage of fatty
streak formation. The majority of cells are macrophages and
macrophage-derived foam cells, together with a varying quantity of
VSMCs. The number of VSMCs increases with the progression of AS,
and becomes the major cellular component of the fibrous and
atheromatous plaques. Previous research has shown that following
endometrial injury, VSMCs migrate from the media to the intima and
alter their form from a constriction to synthetic type (11). The synthesis and secretion of
extracellular matrix components, including collagen, elastin and
proteoglycan increase, together with changes in the secretion of
active substances, including MMPs and TIMPs. As the only cellular
component of the mammalian arterial blood wall, smooth muscle cells
could not only secrete extracellular matrix, but additionally
produce a variety of active substances, including enzymes which are
able to degrade the ECM. Determining the changes in the levels of
active substances secreted by VSMCs would have significant benefit
in studies of AS.
It is currently considered that there are at least
six major categories of ECM-degrading enzymes. These include
prolidase, serine proteases, cysteine proteases, asparagine
proteases, glycosidases, and MMPs, among which MMP is the most
prominent. MMP exerts potent degradation effects on the ECM, thus
acting as a central enzyme in the regulation of ECM homeostasis.
MMPs may be divided into four different categories according to the
target substrate. These include interstitial collagenase,
gelatinase, matrical collagenase and membrane type
metalloproteinases (12). The
activation of progelatinase (MMP-9) on the cell surface
predominantly functions in the degradation of the local matrix,
which would be conclusive towards the migration and proliferation
of cells. TIMPs are endogenous inhibitors of tissue MMPs, forming
MMP-TIMP complexes at a ratio of 1:1, thus blocking the binding of
MMPs and substrates and acting as a transcriptional regulation
mechanism. TIMPs act to inhibit specific MMPs and it has been
demonstrated that TIMP-1 specifically inhibits the activity of
MMP-9 (13–15). The complex interactions of AS
together with the release of growth factors can affect the MMP/TIMP
balance, resulting in an increase in the MMP/TIMP ratio and an
enhancement of the activities of collagenase, thus promoting the
migration of VSMCs. Identifying the effects of the causative
factors for the secretion of MMP-9 and TIMP-1 by VSMC may
facilitate the understanding of the pathogenesis of coronary heart
disease.
The RAAS system participates in the pathological
process of AS, in particular AngII. AngII may act through the
following hypothesized mechanisms: i) Induction of
vasoconstriction, increased blood pressure, and consequently
secondary induction of AS; ii) promotion of proliferation and
vascular remodeling of the smooth muscle cells, inducing the mRNA
expression and protein synthesis of type I and III collagen
(16); iii) promotion of adhesion
molecules, interleukin-6, monocyte chemotactic factor and other
inflammatory cytokines. These three processes are mediated through
the AT1 receptor. It is therefore presumed that AngII receptor
antagonists have anti-atherosclerosis effects. Additional studies
have suggested that losartan, an AT1 receptor antagonist, can
produce an anti-atherosclerotic effect (17,18,7,8).
This may occur through losartan-mediated inhibition of the
degradation of partial matrices, which prevents the migration and
proliferation of VSMCs from the media to the intima, and ultimately
prevents the formation of AS plaques. The present study therefore
analyzed the effects of losartan and AngII on the expression of
MMP-9 and TIMP-1 secreted by rat VSMCs.
AngII has multiple functions in VSMCs, including
affecting hemodynamics and cell growth, which can lead to the
progression of ischemic coronary events. AngII is produced in the
circulating blood or blood vessel walls, resulting in
vasoconstriction and an increase in blood pressure. Production of
AngII additionally has chronic effects, including the direct action
on VSMCs, which leads to the remodeling of various cardiovascular
tissues, including blood vessels and the heart (19). The impacts of AngII differ towards
different cell types. In cultivation experiments of human and rat
cardiac fibroblast cells, AngII was shown to stimulate the
proliferation of cardiac fibroblasts, significantly decrease the
collagenase secretion activity of cardiac fibroblasts, thus
increasing the mRNA and protein expression of type I and III
collagen. This process was shown to be mediated by the AT1 receptor
(20). AngII was able to promote
endothelial cell apoptosis in a dose-dependent manner through the
combined mediation of AT1 and AT2 receptors (21) and was shown to inhibit VSMC
apoptosis through the AT1 receptor (22). Co-incubation of cultured VSMCs with
1.7 nmol/l AngII promoted the secretion of type I collagen from the
cells, which was reciprocally suppressed by losartan. Hadler-Olsen
et al (23) reported that
VSMCs derived from healthy individuals could secrete bioactive
gelatinase in the culture medium; and this activity depended on the
TIMP-bound proMMP, because it could not be fully activated by
p-aminophenylmercuric acetate.
In the present study, analysis of the gene and
protein expression levels showed that AngII could stimulate rat
VSMCs to secrete MMP-9 in a dose-dependent manner, but had no
effect on TIMP-1. Losartan was shown to inhibit the secretion of
MMP-9 by rat VSMCs, at both the gene and protein levels, and
promote the secretion of TIMP-1 in a concentration-dependent
manner. The combined action of losartan and AngII could therefore
inhibit the secretion of MMP-9 by rat VSMCs at the gene and protein
level and promote the secretion of TIMP-1 in a
concentration-dependent manner. The overall effect of losrartan
with AngII was consistent with that of losartan alone.
Morand-Contant et al (24)
confirmed that AngII activates nuclear factor (NF)-κB in VSMCs
through the AT1 receptor, Px. It was additionally shown that
inhibition of NF-κB resulted in a decrease in expression of MMP-1,
MMP-3 and MMP-9 in VSMCs (25).
Another study showed (26) that
AngII promoted the secretion of MMPs from human VSMCs through NF-κB
signaling, which was blocked by losartan. The results of the
present study were consistent with those of previous studies
regarding the impact of AngII and losartan towards the secretion of
MMP-9 by VSMCs. The occurrence and development of AS, as well as
the rupture of plaques, are processes in which the balance of
ECM-degrading enzymes and endogenous inhibitors is altered
(1), however, in-depth studies of
MMP action are required in order to determine the mechanism. It has
been previously identified that TIMP-1 is the endogenous tissue
inhibitor specific for MMP-9, functioning across the progressive
stages of AS (1). The present
study confirmed that AngII did not affect the secretion of TIMP-1
in VSMCs, while losartan could promote the secretion TIMP-1. It
could therefore be concluded that AngII stimulated VSMCs to secrete
MMP-9, altering the balance of MMP-9 and TIMP-1 to increase the
MMP-9/TIMP-1 ratio. This observed trend towards an increase in
collagenase activity, which promotes the migration and
proliferation of VSMCs and other proinflammatory cytokines towards
the intima and the formation of AS plaques and ultimate plaque
rupture. Losartan could inhibit the AT1 receptor, altering the
balance of MMP-9 and TIMP-1 secretion in VSMCs. The subsequent
increase in the TIMP-1/MMP-9 ratio resulted in inhibition of
collagenase activity, thus producing an anti-AS effect.
In conclusion, the present study showed that AngII
could stimulate VSMCs to secret MMP-9 with no effect on TIMP-1
secretion. Losartan was shown to inhibit AngII via the AT1
receptor, promoting VSMCs to secret TIMP-1, thus inhibiting the
secretion of MMP-9. Changes in the MMP-9/TIMP-1 secretion ratio in
VSMCs may be one of the possible mechanisms for AngII promoting and
losartan counteracting AS.
Acknowledgements
The present study was sponsored by the Key Clinical
Specialty Discipline Construction Program of Fujian (Program of
Vasculocardiology), PRC.
References
|
1
|
Peeters W, Moll FL, Vink A, et al:
Collagenase matrix metalloproteinase-8 expressed in atherosclerotic
carotid plaques is associated with systemic cardiovascular outcome.
Eur Heart J. 32:2314–2325. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siasos G, Tousoulis D, Kioufis S, et al:
Inflammatory mechanisms in atherosclerosis: the impact of matrix
metalloproteinases. Curr Top Med Chem. 12:1132–1148. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Newby AC: Dual role of matrix
metalloproteinases (matrixins) in intimal thickening and
atherosclerotic plaque rupture. Physiol Rev. 85:1–31. 2005.
View Article : Google Scholar
|
|
4
|
Yu YM and Lin HC: Curcumin prevents human
aortic smooth muscle cells migration by inhibiting of MMP-9
expression. Nutr Metab Cardiovasc Dis. 20:125–132. 2010. View Article : Google Scholar
|
|
5
|
Johnson JL, Dwivedi A and Somerville M:
Matrix metalloproteinase (MMP)-3 activates MMP-9 mediated vascular
smooth muscle cell migration and neointima formation in mice.
Arterioscler Thromb Vasc Biol. 31:e35–e44. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
An J, Nakajima T, Kuba K and Kimura A:
Losartan inhibits LPS-induced inflammatory signaling through a
PPARγ-dependent mechanism in human THP-1 macrophages. Hypertens
Res. 33:831–835. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Díez J: Review of the molecular
pharmacology of Losartan and its possible relevance to stroke
prevention in patients with hypertension. Clin Ther. 28:832–848.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hernandez-Trujillo Y1,
Rodriguez-Esparragon F, Macias-Reyes A, Caballero-Hidalgo A and
Rodriguez-Perez JC: Rosiglitazone but not losartan prevents Nrf-2
dependent CD36 gene expression up-regulation in an in vivo
atherosclerosis model. Cardiovasc Diabetol. 7:32008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xu YM, Deepak S, Li GP and Zhao YN: Effect
of angiotensin II type 1 receptor antagonist, losartan on
inflammatory factor in atherosclerotic rabbits. Res Cardiocvasc
Med. 1:127–131. 2013. View Article : Google Scholar
|
|
10
|
Xu HX, Li JJ, Li GS, et al: Decreased
infiltration of macrophages and inhibited activation of nuclear
factor-kappa B in blood vessels: a possible mechanism for the
anti-atherogenic effects of losartan. Acta Cardiol. 62:607–613.
2007. View Article : Google Scholar
|
|
11
|
Luan Z, Chase AJ and Newby AC: Statins
inhibit secretion of metalloproteinases-1, -2, -3, and -9 from
vascular smooth muscle cells and macrophages. Arterioscler Thromb
Vasc Biol. 23:769–775. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Madala SK, Pesce JT, Ramalingam TR, et al:
Matrix metalloproteinase 12-deficiency augments extracellular
matrix degrading metalloproteinases and attenuates IL-13-dependent
fibrosis. J Immunol. 184:3955–3963. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang DC, Ma ST and Tan Y: Imbalance of
matrix metalloproteinases/tissue inhibitor of metalloproteinase-1
and loss of fibronectin expression in patients with congestive
heart failure. Cardiology. 116:133–141. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dabek J, Glogowska-Ligus J and Szadorska
B: Transcription activity of MMP-2 and MMP-9 metalloproteinase
genes and their tissue inhibitor (TIMP-2) in acute coronary
syndrome patients. J Postgrad Med. 59:115–120. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ding SF, Liu HJ, Lu Q, et al: Changes of
matrix metalloproteinase-9 and tissue inhibitors of
metalloproteinase-1 during left ventricular remodeling in acute
myocardial infarction patients after percutaneous coronary
intervention. Biomed Res-India. 24:179–184. 2013.
|
|
16
|
Virdis A, Duranti E and Taddei S:
Oxidative stress and vascular damage n hypertension: Role of
angiotensin II. Int J Hypertens. 2011.Epub, 2011. View Article : Google Scholar
|
|
17
|
Lee BS, Choi JY, Kim JY, et al:
Simvastatin and losartan differentially and synergistically inhibit
atherosclerosis in apolipoprotein e(−/−) mice. Korean Circ J.
42:543–550. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kaynar K, Ulusoy S, Ovali E, et al:
TGF-beta and TNF-alpha producing effects of losartan and amlodipine
on human mononuclear cell culture. Nephrology (Carlton).
10:478–482. 2005. View Article : Google Scholar
|
|
19
|
Yaghooti H, Firoozrai M, Fallah S and
Khorramizadeh MR: Angiotensin II differentially induces matrix
metalloproteinase-9 and tissue inhibitor of metalloproteinase-1
production and disturbs MMP/TIMP balance. Avicenna J Med
Biotechnol. 2:79–85. 2010.PubMed/NCBI
|
|
20
|
Gu J, Liu X, Wang QX, et al: Angiotensin
II increases CTGF expression via MAPKs/TGF-β1/TRAF6 pathway in
atrial fibroblasts. Exp Cell Res. 318:2105–2015. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
O’Reilly MA: Angiotensin II: tapping the
cell cycle machinery to kill endothelial cells. Am J Physiol Lung
Cell Mol Physiol. 303:L575–L576. 2012. View Article : Google Scholar :
|
|
22
|
deBlois D, Tea BS, Than VD, Tremblay J and
Hamet P: Smooth muscle apoptosis during vascular regression in
spontaneously hypertensive rats. Hypertension. 29:340–349. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hadler-Olsen E, Fadnes B, Sylte I,
Uhlin-Hansen L and Winberg JO: Regulation of matrix
metalloproteinase activity in health and disease. FEBS J.
278:28–45. 2011. View Article : Google Scholar
|
|
24
|
Morand-Contant M, Anand-Srivastava MB and
Couture R: Kinin B1 receptor upregulation by angiotensin II and
endothelin-1 in rat vascular smooth muscle cells: receptors and
mechanisms. Am J Physiol Heart Circ Physiol. 299:H1625–H1632. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pan P, Fu H, Zhang L, et al: Angiotensin
II upregulates the expression of placental growth factor in human
vascular endothelial cells and smooth muscle cells. BMC Cell Biol.
11:362010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kranzhoeter R and Larsen D: Angiotensin II
induces relaease of matrix metalloproteinases from human vascular
smooth muscle cells via NF-kappa B. Circulation. 102:11–12.
2000.
|














