Introduction
Endometrial carcinoma is a common malignancy of the
female genital tract. The incidence of endometrial carcinoma has
increased in recent years, and it has become the most common
gynecological malignancy in a number of European and American
countries (1). By ruling out
interference from various confounding factors with laser capture
microdissection technology, cyclin-dependent kinase (CDK) 7 has
been identified as a differentially expressed gene that is highly
correlated with endometrial carcinoma (2,3). As
a basic structural component of CDK-activating kinase (CAK), CDK7
has a key role in the cell cycle. Abnormal expression of CDK7
disturbs the balance of the cell cycle and promotes DNA replication
and mitosis, resulting in abnormal cell growth, replication, and
differentiation; abnormalities that are closely associated with the
occurrence and development of tumors. Additional research following
these studies revealed that the expression levels of CDK7 were
lowest in normal endometrium, increased during endometrial
hyperplasia and peaked in endometrial carcinoma tissues (4).
Based on preliminary research, the present study
used siRNA technology to silence CDK7 expression in the HEC-1-A
endometrial carcinoma cell line. Changes in the cisplatin
[cis-dichlorodiammineplatinum (II), or DDP] sensitivity of cells
were detected by MTT cytotoxicity assay in addition to flow
cytometry and Hoechst/propidium iodide (PI) co-immunofluorescence
microscopy. The aims of the present study were to clarify the
association between CDK7 expression levels and the sensitivity of
HEC-1-A cells to cisplatin, and to reveal the mechanism underlying
chemotherapy resistance in endometrial carcinoma cells, providing a
novel theoretical basis for the improvement of chemotherapy
efficacy. To the best of our knowledge, no similar studies have
been performed either domestically or internationally.
Materials and methods
Human endometrial carcinoma cell
culture
HEC-1-A cells were purchased from the Tianjin
Institute of Hematology (Tianjin, China) and were routinely
cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco-BRL,
Carlsbad, CA, USA) containing 10% fetal bovine serum (FBS). The
cells were maintained in a humidified incubator at 37°C with 5%
CO2.
Design and synthesis of CDK-7 siRNA
fragments
GenePharma siRNA designing software was used to
design the siRNA fragments (GenePharma, Shanghai, China). Based on
the gene sequence of human CDK7, four different siRNA fragments,
including CDK7-1, CDK7-2, CDK7-423 and CDK7-910, were designed and
synthesized by Shanghai UNJA Biotechnology, Ltd. (Shanghai,
China).
Transfection of siRNA
HEC-1-A cells were grown to 80–90% confluence and
subsequently inoculated onto 6-well plates at a density of
8×105 cells/well. Once they had been mixed thoroughly,
the cells were cultured at 37°C in a 5% CO2 incubator
for 24 h. The cells were divided into six groups: CDK7-1, CDK7-2,
CDK7-423, CDK7-910, the negative control (with siRNA constructed
from unrelated sequences), and the blank control group (normal
cultured cells). Opti-MEM serum-free culture medium (250 μl;
Gibco-BRL) and 100 pmol siRNA were added to a 1.5 ml Eppendorf (EP)
tube, while 250 μl of Opti-MEM and 5 μl of Lipofectamine™ 2000
(Gibco-BRL) were added into a second EP tube. Following gentle
mixing, the solutions were placed at room temperature for 5 min,
gently mixed again and incubated at room temperature for 20 min.
Subsequently, the culture medium was removed, and 1.5 μl of
Opti-MEM was added to each well. The transfection mixture was added
dropwise to 6-well plates, and the cells were incubated for 4–6 h.
Following incubation, the transfection solution was discarded, and
500 μl of DMEM culture medium containing 10% FBS was added. The
cells were cultured at 37°C in 5% CO2 for 48 h and
harvested for RNA extraction or western blot analysis. The
transfection efficiency = the number of GFP-labeled cells/total
number of cells × 100, where GFP is green fluorescence protein.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
The total RNA in the transfected cells was extracted
using TRIzol® (Invitrogen, Carlsbad, CA, USA) following
the manufacturer’s instructions. cDNA was then synthesized and the
qPCR used the following primers: CDK7 upstream,
5′-AGGATGTATGGTGTAGGTGTGGA-3′, and downstream,
5′-AAGATGTGATGCAAAGGTATTCC-3′ (amplification length, 221 bp) and
GAPDH upstream, 5′-AGAAGGCTGGGGCTCATTTC-3, ′ and downstream
5′-AGGGGCCATCCACAGTCTTC-3′ (amplification length, 220 bp). The
primers were designed using the Primer Premier 5.0 software
(PREMIER Biosoft, Palo Alto, CA, USA) and were synthesized by
Shanghai Sangon Biological Technology Co., Ltd. (Shanghai, China).
The cycling conditions for the reverse transcription were as
follows: 70°C for 5 min, followed by immediate cooling on ice; 42°C
for 30 min; and 85°C for 10 min for reaction termination, followed
by immediate cooling on ice. cDNA synthesis was conducted using a
MuLV reverse transcriptase kit (Applied Biosystems, Foster City,
CA, USA) according to the manufacturer’s instruction. The cDNA
reaction solution was used as a template for the subsequent step.
qPCR was performed using an Applied Biosystems 7500 Real-Time PCR
Machine and data were analyzed using Step One Software v.2.1
(Applied Biosystems). GAPDH was used as an internal normalization
control. The results are represented as the fold change in gene
expression relative to that of GAPDH (2−ΔΔCt). The
primers and probes chosen from Roche’s UPL system were as follows:
CDK7 (accession no: NM_000077.4) with UPL probe #34, and GAPDH
(accession no: NM_0000194.4) with UPL probe #73. The reaction
conditions for the quantitative PCR were as follows: 95°C for 2
min; followed by 40 cycles of 95°C for 20 sec, 60°C for 30 sec and
72°C for 30 sec; and finally a 72°C extension for 10 min.
Western blot analysis
A total of 100 μl cell lysis buffer (Beyotime,
Shanghai, China). was added to each well of cells. The lysate was
then transferred into a centrifuge tube and heated to 100°C for 5
min. Following cooling on ice, the sample was centrifuged at 12,000
× g for 10 min to remove any insoluble precipitate. Subsequently,
the harvested sample was separated using 10% SDS-PAGE (Beijing
Biyutian Co., Ltd., Beijing, China). Following electrophoresis, the
sample was transferred onto a polyvinylidene fluoride membrane
(Shanghai ShuoGuang Technology Co., Ltd., Shanghai, China) and
blocked using 5% skim milk (Gibco-BRL). The membrane was incubated
with anti-CDK7 antibodies (1:500, Abcam, Cambridge, UK) at 4°C
overnight. Subsequently, the cells were incubated with horseradish
peroxidase-labeled rabbit anti-goat IgG (ZSGB-BIO, Beijing, China)
at an appropriate dilution at room temperature for 2 h.
Chemiluminescence detection was performed using the enhanced
chemiluminescence (ECL) reagent (Roche, Basel, Switzerland) and
exposed to ECL X-ray films (Roche). After being developed and
fixed, images of the films were captured using a gel imaging
analysis system (BioRad Laboratories, Hercules, CA, USA ). The
results were analyzed using the Gel-Pro-Analyzer (Media
Cybernetics, Georgia, MD, USA). GADPH was used as the internal
control and the experiments were repeated three times.
MTT cytotoxicity assay
HEC-1-A cells at the logarithmic growth phase were
transfected with CDK7-423. After 48 h, the cells were dissociated,
harvested and inoculated onto a 96-well plate, with 7,000–8,000
cells/well. Once the cells attached, they were treated with 0.4,
2.0, 10.0, 50.0 or 250.0 μg/ml cisplatin (Qilu Pharmaceutical Co.,
Shandong, China) and incubated at 37°C in 5% CO2 for 48
h. Subsequently, 10 μl of MTT (5 mg/ml; Shanghai Yuanye Biological
Technology Co., Ltd., Shanghai, China.) was added to each well, and
the plates were incubated at 37°C for 4 h. The culture plates were
removed from the incubator and centrifuged at 550 × g for 5 min.
The supernatant was discarded and 100 μl dimethylsulfoxide
(Shanghai Yuanye Biological Technology Co., Ltd.) was added to each
well to terminate the reaction and dissolve the purple/blue
formazan. The mixtures were vortexed and each well’s absorbance
value was detected using a μQuant microplate reader (Bio-Tek
Instruments Inc., Winooski, VT, USA) at a wavelength of 490 nm
wavelength. The inhibition rates of the cisplatin-treated HEC-1-A
cells were calculated prior to and following CDK7 siRNA
transfection. The calculated rates were then used for curve fitting
and half maximal inhibitory concentration (IC50)
calculations.
Analysis of cell cycle and detection of
apoptosis rate using flow cytometry
HEC-1-A cells were counted prior to and after CDK7
siRNA transfection and adjusted to a final concentration of
1×106 cells/ml. Subsequently, 2 ml of cells were
inoculated onto a 6-well plate. Following treatment with cisplatin
(10 μg/ml) for 48 h, the cells were dissociated, collected and
stained with PI. The cells were detected and analyzed using an
Elite flow cytometer (Coulter Cytometry, Inc., Hialeah, FL,
USA).
Observation of nucleus morphological
changes using immunofluorescence microscopy
HEC-1-A cells were counted prior to and after CDK7
siRNA transfection, and the cell concentration was adjusted to
7–8×104 per ml. Subsequently, 1 ml of cells was
inoculated onto a 24-well plate and treated with cisplatin (10
μg/ml) for 48 h. A staining solution of Hoechst 33258 in
phosphate-buffered saline was added and incubated at 37°C for 15
min. PI dissolved in PBS (10 g/ml) was added at room temperature
for 15 min to cause a reaction. Images of the cells were captured
under a fluorescence microscope (Leica, Mukwonago, WI, USA).
Statistical methods
The χ2 test was performed using SPSS
version 13.0 software (SPSS, Inc., Chicago, IL, USA). Comparisons
between the means of two groups were performed using an independent
groups t-test. Comparisons of the means among multiple samples were
performed using single-factor analysis of variance (ANOVA). An
analysis of the time- and dose-dependent responses was performed
using an ANOVA of factorial design. P<0.05 was considered to
indicate a statistically significant difference.
Results
Transfection efficiency of CDK7 siRNA
increases in a time-dependent manner
CDK7 siRNA was labeled with GFP and transfected into
HEC-1-A cells. The results showed that the transfection efficiency
at 48 h was significantly higher than that at 24 h, reaching ~70%
(Fig. 1).
CDK7 siRNA inhibits CDK7 expression
levels in HEC-1-A cells
The total RNA in CDK7-transfected cells was
extracted using the TRIzol® method. The obtained RNA was
dissolved in diethyl phosphorocyanidate (DEPC)-treated water, and
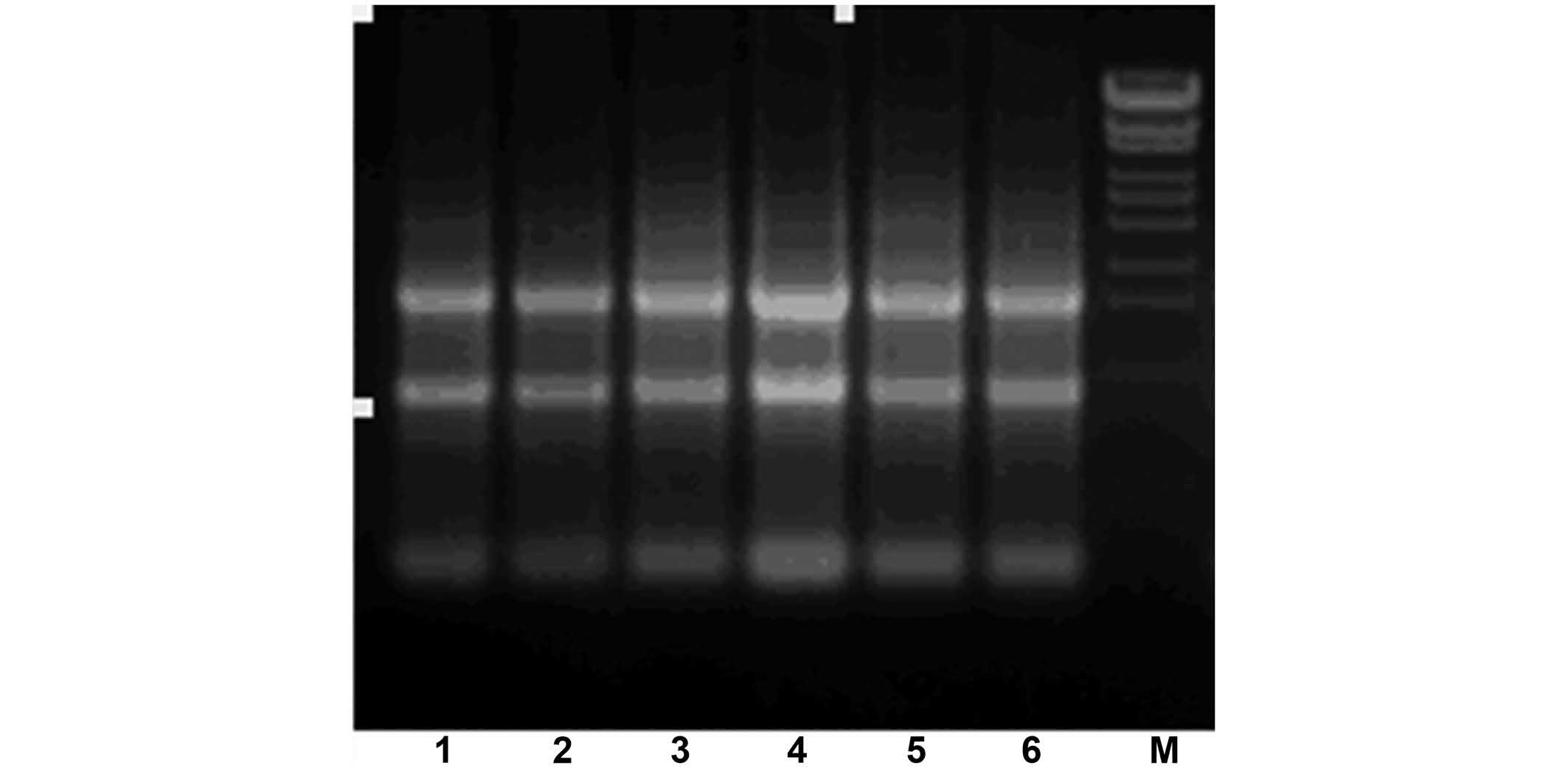
the integrity of the RNA was analyzed using agarose gel
electrophoresis (Fig. 2). The PCR
amplification curves, standard curves and dissociation curves of
CDK7 and GADPH were satisfactory. A comparison revealed that the
interference effect of CDK7-423 (group 3) was the strongest
(Table I). The results of the
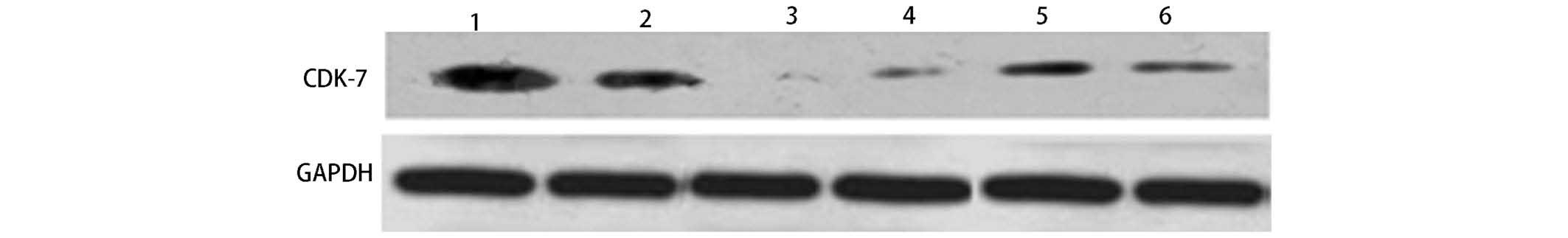
western blotting revealed that the siRNA interference in each group
reduced the CDK7 protein expression levels. The interference effect
of CDK7-423 was the strongest, which corresponded to the results of
the western blotting (Fig. 3).
 | Table ICt values of CDK7 and GAPDH |
Table I
Ct values of CDK7 and GAPDH
| Identifier | CDK7 (Ct) | GAPDH (Ct) |
|---|
| 1 | 17.57±0.21 | 11.77±0.19 |
| 2 | 18.14±0.35 | 12.20±0.08 |
| 3 | 18.74±0.32 | 12.06±0.12 |
| 4 | 18.45±0.18 | 12.31±0.21 |
| 5 | 17.34±0.19 | 12.65±0.24 |
| 6 | 17.62±0.38 | 12.82±0.33 |
Suppression of CDK7 expression in HEC-1-A
cells induces significantly higher cisplatin cytotoxicity
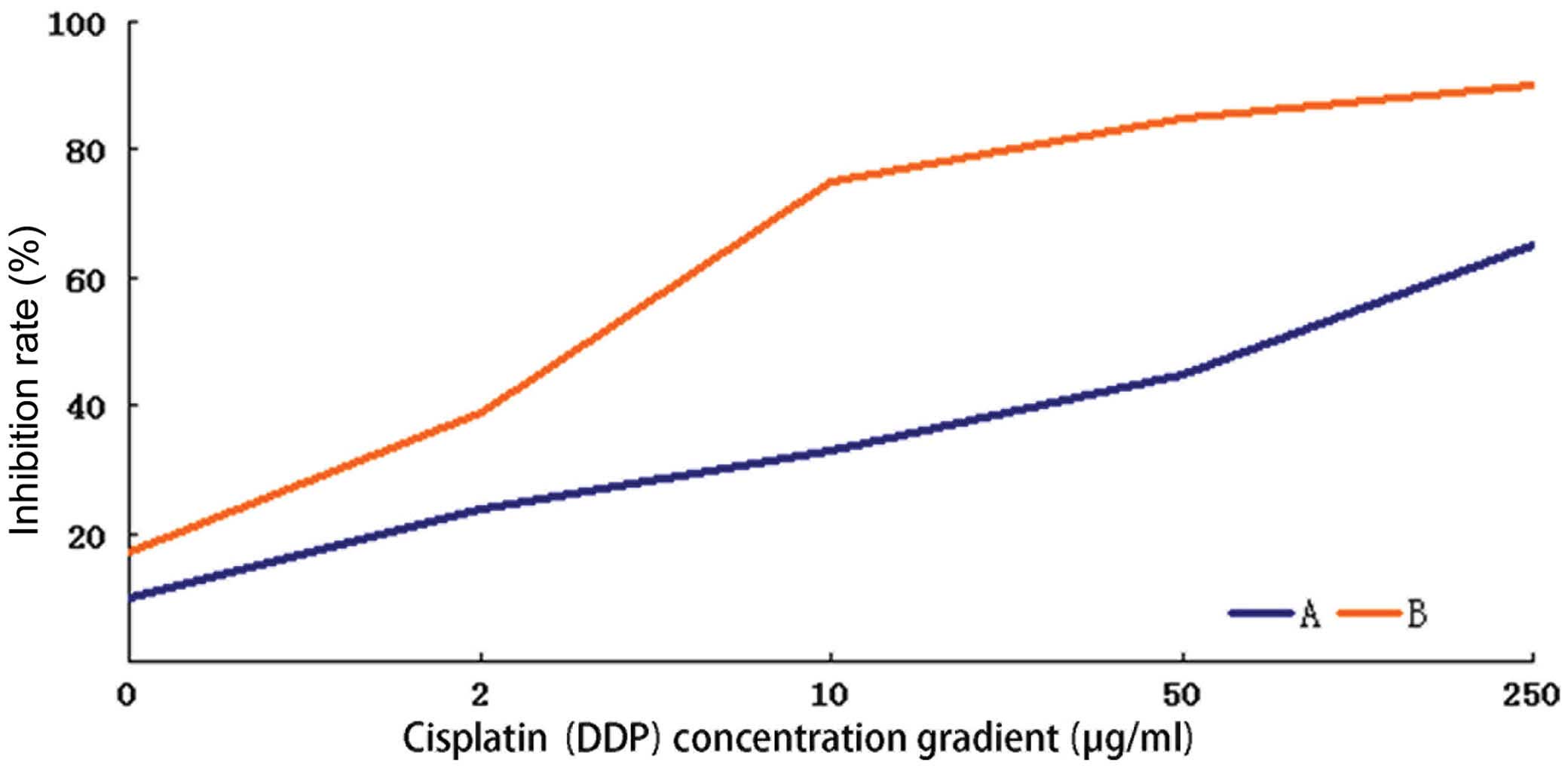
The inhibition rates induced by 48-h treatment with
different concentrations of cisplatin were calculated in the
HEC-1-A cells before and after CDK7-423 siRNA transfection, and a
fitted curve was obtained for the determination of IC50.
The IC50 of cisplatin was 45.122 μg/ml in the parental
HEC-1-A cells, which reduced to 3.200 μg/ml following transfection
with CDK7-423 siRNA. The concentration gradient of cisplatin used
in this experiment and the corresponding inhibition rates are shown
in Fig. 4. The 48-h cisplatin
treatment induced significantly higher cytotoxicity in the HEC-1-A
cells with inhibited CDK7 expression, compared with that observed
in the parental cells (P<0.05).
CDK7 knockdown by siRNA increases the
apoptosis rates in cisplatin-treated HEC-1-A cells
Following the 48-h cisplatin treatment, at a final
concentration of 10 μg/ml, the mean apoptosis rates were 11.66% in
the parental HEC-1-A cells and 37.57% in the cells transfected with
CDK7-423 siRNA (P<0.05) (Fig.
5).
Cisplatin treatment significantly
increases the number of apoptotic bodies in HEC-1-A cells with low
CDK7 expression levels
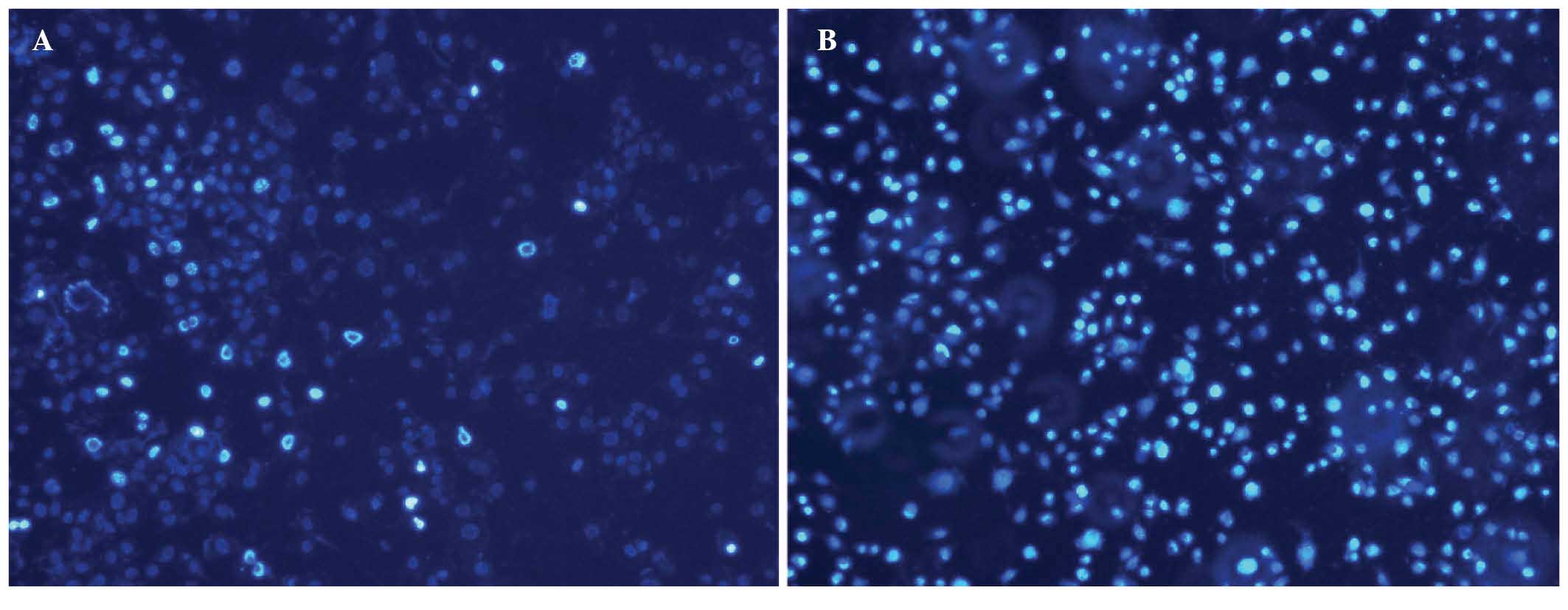
Following treatment with 10 μg/ml cisplatin for 48
h, observations under the immunofluorescence microscope showed that
the number of apoptotic bodies (bright aggregates or
snowflake-shaped fluorescent spots that are the characteristic
spots of apoptotic nuclei, caused by chromatin condensation) in
HEC-1-A cells transfected with CDK7-423 siRNA significantly
increased, when compared with those observed in the parental cells
(P<0.05) (Fig. 6).
Discussion
CDKs belong to the serine/threonine kinase family.
Currently, nine CDK family members (CDK1-CDK9) have been discovered
in mammals. By binding to different cyclins to form complexes,
these CDKs directly or indirectly act on the different phases of
the cell cycle to maintain normal cell growth, differentiation and
proliferation. Disturbances in the cell cycle may result in
persistent cell growth and eventually tumor occurrence (5). Unlike other CDK family members (CDKs
1–4 and 6) that are directly involved in the cell cycle, CDK7
primarily participates in the regulatory processes of the cell
cycle. As an important component of CAK, CDK7 has a pivotal role in
cell cycle regulation. CAK is a complex consisting of three
subunits: CDK7, cyclin H and MAT1. Of these subunits, CDK7 is the
catalytic subunit and cyclin H is the regulatory subunit. CDK7
phosphorylation activates CAK, resulting in the phosphorylation and
activation of the CDK molecules that bind to mitotic-type cyclins
(including, cyclin A and B), thereby stimulating cells to enter the
M phase from the G2 phase. In addition, CDK7
phosphorylation-induced CAK activation phosphorylates and activates
CDK molecules, which bind to G1-phase cyclins
(including, cyclin D and E) and promote cells to enter the S phase
from the G1 phase, thus encouraging cell division and
proliferation (6). Additionally,
this complex is an important component of the basic transcription
factor TFIIH. TFIIH catalyzes the phosphorylation of the large
subunit of RNA polymerase II to trigger the transcription process.
It also participates in type II transcription and nucleotide
excision repair to prolong the transcription phase; as a result,
genes that participate in cell division and proliferation are
expressed, again promoting cell proliferation (7).
Due to its regulatory effect on the activity of
other CDKs, CAK has a key role in the process of cell cycle
regulation. As an important component of CAK, CDK7 is a promising
therapeutic target for a variety of anti-carcinoma chemotherapeutic
regimens. Previous studies have reported that the silencing of CDK7
expression via a number of methods suppresses the growth and
proliferation of liver carcinoma, lymphoma, leukemia, intestinal
carcinoma and breast cancer cells (8–12).
Using the structure of CDK7 as a starting point, Liu et al
(13) established a molecular
docking model of CDK7 inhibitors and synthesized a novel CDK7
inhibitor; their results showed that this novel compound had
inhibitory effects on HL60 acute promyelocytic leukemia cells, KB
nasopharyngeal carcinoma cells, SMMC-7721 liver carcinoma cells,
HCT-116 colon adenocarcinoma cells and A549 lung carcinoma cells.
Therefore, CDK7 was hypothesized to be a novel target for a variety
of anti-carcinoma drug treatments.
Among the various clinical treatment measures,
chemotherapy has an important role in the comprehensive treatment
of endometrial carcinoma. Postoperative chemotherapy is essential
for the eradication of residual tumor cells. Chemotherapy is
becoming the first-line treatment for advanced-stage cancer
patients with small residual lesions and early-stage high-risk
cancer patients; it is the primary treatment method for advanced
and recurrent endometrial carcinomas. Platinum-based drugs (such as
cisplatin and carboplatin) are the most widely used chemotherapy
drugs in endometrial carcinoma; however, the efficacy of
endometrial carcinoma chemotherapy is not satisfactory. Previous
studies have reported that theefficacy of cisplatin alone is ~30%;
and whilst combined chemotherapy may have increased efficacy, the
toxic side-effects increase accordingly (14–16).
Chemotherapy resistance causes cancer treatments to be ineffective,
resulting in enormous physical, psychological and economic losses
to patients. Therefore, developing methods to increase chemotherapy
sensitivity (to agents including cisplatin) and overcome drug
resistance has become a research hotspot in the clinical treatment
of endometrial carcinoma.
Yang (17) used
gene chip technology to screen the differentially expressed genes
in cisplatin-resistant lung adenocarcinoma cells and found that
CDK7 was highly expressed, suggesting that CDK7 is associated with
cisplatin resistance in lung carcinomas. RNA interference
technology was used to specifically silence CDK7 and observe the
effect of CDK7 downregulation on the biological characteristics of
cisplatin-resistant A549/CDDP human lung carcinoma cells. The
results revealed that in lung adenocarcinoma, CDK7 is involved in
the development of cisplatin resistance. In addition to its effect
on cell cycle regulation, CDK7 may also mediate cisplatin
resistance via the drug resistance-associated protein pathway.
However, RNA interference may partially reverse the CDK7-mediated
drug resistance in lung adenocarcinoma cells. Therefore, it is
possible that CDK7 may be used as a gene therapy target for
chemotherapy resistance in lung adenocarcinoma.
Based on the previous studies described, the current
study focused on CDK7 as a research target. To the best of our
knowledge, no studies on silencing CDK7 in endometrial carcinoma
cells using siRNA technology have been reported, either
domestically or internationally. In the present study, four
different siRNA fragments were designed based on the sequence of
the CDK7 gene. These were successfully transfected into the
endometrial carcinoma cell line HEC-1-A.
The results of RT-qPCR and western blotting
indicated that each type of interfering RNA suppressed the levels
of CDK7 protein expression to varying degrees. The RNA interference
mediated by CDK7-423 was the strongest, inhibiting >70% of the
protein expression compared with that of the controls. To reveal
the association between CDK7 and platinum resistance in endometrial
carcinoma cells, CDK7-423 was selected to specifically reduce CDK7
expression in HEC-1-A endometrial carcinoma cells, and the MTT
cytotoxicity assay, flow cytometry and Hoechst/PI double-staining
immunofluorescence microscopy were used to detect changes in
cisplatin sensitivity. The results showed that following 48-h
cisplatin treatment, the IC50 of cisplatin was 45.122
μg/ml in the parental HEC-1-A cells, while it was only 3.200 μg/ml
in the CDK7 low-expression group, indicating a statistically
significant higher cytotoxicity in the cells with low CDK7
expression than in the parental cells (P<0.05). Following the
48-h cisplatin (10 μg/ml) treatment, the average apoptosis rate was
11.66% in parental HEC-1-A cells, which increased to 37.57% in the
CDK low-expression group (P<0.05). Compared with the parental
HEC-1-A cell group, the number of apoptotic cells in the CDK7
low-expression group was significantly increased, as observed under
a fluorescence microscope. These results indicate that following
the suppression of CDK7 expression levels in endometrial carcinoma
cells, the sensitivity of the cells to the chemotherapy drug
cisplatin was significantly increased. Thus, high CDK7 expression
may be one of the mechanisms underlying the resistance of
endometrial carcinoma cells to platinum-based chemotherapy.
In conclusion, the results of the present study may
provide novel ideas and a theoretical basis to improve the clinical
efficacy of chemotherapy and to reverse chemotherapy resistance.
Further in-depth studies using CDK7 as a target for endometrial
carcinoma treatment should be performed.
References
|
1
|
Moxley KM and McMeekin DS: Endometrial
carcinoma: a review of chemotherapy, drug resistance, and the
search for new agents. Oncologist. 15:1026–1033. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wen-Xin L and Xi-Shan H: Application of
laser capture microdissection and differential display technique
for screening of pathogenic genes involved in endometrial
carcinoma. Int J Gynecol Cancer. 17:1224–1230. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu WX and Hao XS: Screening, cloning and
identification of the human endometrial carcinoma-related genes.
Zhonghua Zhong Liu Za Zhi. 29:584–588. 2007.(In Chinese).
|
|
4
|
Liu WX, Liu YX and Chen Y: Expression and
significance of CDK7 and CyclinH in endometrial carcinoma. Shandong
Medical Journal. 51:74–75. 2011.(In Chinese).
|
|
5
|
Zhan SS, Yuan W and Cai JY: Dependent
kinase activation cell cycle protein kinase and tumor. China
Practical Medicine. 5:243–244. 2010.
|
|
6
|
Suryadinata R, Sadowski M and Sarcevic B:
Control of cell cycle progression by phosphorylation of
cyclin-dependent kinase (CDK) substrates. Biosci Rep. 30:243–255.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhovmer A, Oksenych V and Coin F: Two
sides of the same coin: TFIIH complexes in transcription and DNA
repair. Scientific World Journal. 13:633–643. 2010. View Article : Google Scholar
|
|
8
|
Zhao AG and Wu SG: Silence CDK7 and CDK2
leads to phosphorylation of pRb and induces apoptosis in HepG2
cells decreased. Chinese Pharmacological Bulletin. 21:106–110.
2005.(In Chinese).
|
|
9
|
Tong WG, Chen R, Plunkett W, Siegel D,
Sinha R, Harvey RD, Badros AZ, Popplewell L, Coutre S, Fox JA, et
al: Phase I and pharmacologic study of SNS-032, a potent and
selective Cdk2, 7, and 9 inhibitor, in patients with advanced
chronic lymphocytic leukemia and multiple myeloma. J Clin Oncol.
28:3015–3022. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Boquoi A, Chen T and Enders GH:
Chemoprevention of mouse intestinal tumorigenesis by the
cyclin-dependent kinase inhibitor SNS-032. Cancer Prev Res.
2:800–806. 2009. View Article : Google Scholar
|
|
11
|
Walsby E, Lazenby M, Pepper C and Burnett
AK: The cyclin-dependent kinase inhibitor SNS-032 has single agent
activity in AML cells and is highly synergistic with cytarabine.
Leukemia. 25:411–419. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dickson MA and Schwartz GK: Development of
cell-cycle inhibitors for cancer therapy. Curr Oncol. 16:36–43.
2009.PubMed/NCBI
|
|
13
|
Liu T, Sun MT, Dong XW, Ren X, Yang X, Du
LL and Hu YZ: Structure-Based Drug Design, Synthesis and Antitumor
Activities of Novel CDK7 Inhibitors. Acta Phys Chim Sin.
25:2107–2112. 2009.(In Chinese).
|
|
14
|
Moxley KM and McMeekin DS: Endometrial
carcinoma: a review of chemotherapy, drug resistance, and the
search for new agents. Oncologist. 15:1026–1033. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Humber CE, Tiemey JF, Symonds RP, et al:
Chemotherapy for advanced, recurrent or metastatic endometrial
cancer: a systematic review of Cochrane collaboration. Ann Oncol.
18:409–420. 2007. View Article : Google Scholar
|
|
16
|
Shen XY and Xiang Y: Endometrial cancer
chemotherapy. International Journal of Obstetrics and Gynecology.
37:436–439. 2010.
|
|
17
|
Yang HZ: Cisplatin resistance lung
adenocarcinoma cancer screening of differentially expressed genes
and functional. unpublished PhD thesis. Central South University;
2007
|