Introduction
Non-alcoholic fatty liver disease (NAFLD) is the
most common type of liver disease in the general population
(1). NAFLD includes simple
steatosis, non-alcoholic steatohepatitis (NASH), cirrhosis and
hepatocellular carcinoma (HCC) (2). Although NAFLD is benign, it has been
reported that 20% of patients with NAFLD progress to NASH,
cirrhosis and HCC (3,4). The pathophysiological events and
effective therapies for NASH remain to be elucidated.
Previous clinical studies have reported that
endotoxin/toll-like receptor 4 (TLR4) signaling is crucial in the
activation of inflammatory pathways associated with NASH (5). TLR4 is a pattern recognition
receptor, which recognizes endotoxin and signals through adaptor
molecules, termed myeloid differentiation primary response gene 88
and Toll/interleukin-1 receptor domain-containing adaptor-inducing
interferon-β, to activate transcription factors that initiate
innate immunity (6). TLR4 is
expressed in multiple liver cell types, including liver vascular
endothelial cells, Kupffer cells and hepatic stellate cells (HSC)
(7,8). The effect of TLR4 on HSC is integral
to fibrosis development through its effects on transforming growth
factor-β (TGF-β)-dependent collagen production (8).
In human and animal studies, it has been reported
that NASH is associated with portal LPS levels through mechanisms
involving bacterial translocation (9,10)
and the gut microbiota is considered to generate products such as
lipopolysaccharide (LPS), a cell-wall component of Gram-negative
bacteria, which is delivered into the liver via the portal vein
(11,12). Endotoxin production by gut
microbiota may cause inflammation in patients with obesity,
diabetes, metabolic disorder, NAFLD and NASH (11,13).
Plasma LPS levels are associated with small intestinal bacteria
overgrowth, the change of composition of the microbiota and
increased intestinal permeability (14).
Polymyxins are antibiotics with a structure
consisting of a cyclic peptide with a long hydrophobic tail. They
are selectively toxic to Gram-negative bacteria, including
Escherichia coli, Pseudomonas aeruginosa, Klebsiella
pneumoniae and other members of the Enterobacteriaceae family
due to their specificity for the LPS molecule, which exists within
a number of Gram-negative outer membranes. They are produced by
nonribosomal peptide synthetase systems in Gram-positive bacteria,
including Paenibacillus polymyxa and disrupt the structure
of the bacterial cell membrane by interacting with phospholipids.
They are not absorbed through the gastrointestinal tract. In
clinical settings, they are used for patients with Gram-negative
bacterial infections and for endotoxin apheresis column treatment
of endotoxemia (15).
Neomycins are aminoglycoside antibiotics and are
effective against Gram-negative and Gram-positive bacteria. They
are produced by Gram-positive bacteria, including Streptomyces
fradiae. They inhibit the protein synthesis of bacteria via
binding to 30S ribosomes. Their absorption through the
gastrointestinal tract is limited and they are useful for
Gram-negative bacterial infections in clinical settings.
In the present study, the effects of these poorly
absorbed antibiotics on intestinal permeability and on the
progression of liver fibrosis were assessed. The results revealed
that, in a rat model of choline deficient amino acid-induced liver
fibrosis, the administration of poorly absorbed antibiotics led to
reduced intestinal permeability and decreased liver fibrosis.
Consequently, the present study elucidated the role of LPS in the
pathogenesis of NASH.
Materials and methods
Animal model of liver disease
Male six-week-old Fischer 344 rats (CLEA Japan,
Inc., Osaka, Japan) were housed in a room under controlled
temperature and a 12/12 h light-dark cycle. The animals were
divided into the following three experimental groups and fed for 8
weeks: i) Choline-deficient amino acid diet (CDAA; n=10); ii)
choline-deficient amino acid diet plus antibiotics (CDAA+AB; n=10)
and iii) choline-supplemented amino acid diet (CSAA; n=5). All rats
were sacrificed at the end of week 8. For selective intestinal
decontamination, poorly absorbable antibiotics (1 g/l polymyxin B
sulfate salt (Fluka, Buchs, Switzerland) and 3 g/l neomycin
trisulfate salt hydrate (Sigma-Aldrich, St. Louis, MO, USA) were
administered to the rats in the CDAA+AB group by adding them to
drinking water during the experimental period, excluding the first
and fifth week. All animal procedures were performed according to
standard protocols and in accordance with the standard
recommendations for the proper care and use of laboratory animals.
This study was approved by the ethics committee of Nara Medical
University, Kashihara, Japan.
Histological examination
Conventional histological examination was performed
using hematoxylin and eosin and Sirius-red (Narabyouri Research,
Nara, Japan) staining of the excised liver sections, as described
previously (16).
Immunohistochemistry
For immunostaining of the α-smooth muscle actin
(α-SMA), 5 μm-thick liver sections were stained using the indirect
immunoperoxidase technique with mouse anti-human monoclonal
anti-α-SMA antibody (undiluted; Dako Japan, Co., Ltd., Kyoto,
Japan), as described previously (16). For the immunofluorescence
examination, frozen liver and intestinal sections were fixed with
4% paraformaldehyde for 10 min at 4°C and blocked with 3% bovine
serum albumin for 1 h at room temperature to eliminate background
staining. The tissue sections were then incubated with primary
antibodies rabbit anti-rat polyclonal zona occludens antibody
(ZO-1; 1:100; Invitrogen Life Technologies, Carlsbad, CA, USA) and
mouse anti-rat monoclonal Claudin-4 antibody (1:100; Invitrogen
Life Technologies) at 4°C overnight. This was followed by
incubation with the appropriate donkey anti-rabbit Alexa Fluor-488
or goat anti-mouse Alexa Fluor-546 secondary antibodies (1:200;
Invitrogen Life Technologies) for 1 h at room temperature. The
nuclei were counterstained with 4′,6-diamidino-2-phenylindole
Fluoromount-G (Southern Biotech, Birmingham, AL, USA). The
immunofluorescent staining was visualized using a Zeiss Axiovert 40
CEL® microscope (Zeiss, Jena, Germany) and images from
ZO-1 and Claudin-4 staining were quantified using Axio software
version 4® (Zeiss). For quantification, five images were
randomly selected for quantification analysis from each sample and
the software program quantified the staining intensity of the
selected images based on a preselected threshold.
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the liver and
intestinal tissue samples using acid guanidinium
thiocyanate-phenol-chloroform extraction. The mRNA levels of
collagen Iα, TGF-β, TLR4 and LPS-binding protein (LBP) in the liver
and TLR4 in the intestine were measured by qPCR using the Applied
Biosystems StepOnePlus™ Real-Time PCR® (Applied
Biosystems, Foster City, CA, USA), as described previously
(17). Primer sequences were as
follows: β-actin, forward 5′-GGA GAT TAC TGC CCT GGC TCC TA-3′ and
reverse 5′-GAC TCA TCG TAC TCC TGC TTG CTG-3′; TLR4, forward 5′-CCG
CTC TGG CAT CAT CTT CA-3′ and reverse 5′-CCC ACT CGA GGT AGG TGT
TTC TG-3′; LBP, forward 5′-AAC ATC CGG CTG AAC ACC AAG-3′ and
reverse 5′-CAA GGA CAG ATT CCC AGG ACT GA-3′; TGF-β, forward 5′-CGG
CAG CTG TAC ATT GAC TT-3′ and reverse 5′-AGC GCA CGA TCA TGT TGG
AC-3′ and collagen Iα, forward 5′-AGC TCC TGG GCC TAT CTG ATG A-3′
and reverse 5′-AAT GGT GCT CTG AAA CCC TGA TG-3′. The cycling
conditions were as follows: Initial holding stage at 95°C for 20
sec; followed by 40 cycles of 95°C for 3 sec and 60°C for 30 sec;
followed by the melting curve stage of 95°C for 15 sec, 60°C for 1
min and 95°C for 15 sec.
Protein expression analysis
The hepatic tissue was homogenized in lysis buffer
(Tissue Protein Extraction Reagent; Thermo Scientific, Kanagawa,
Japan) containing a mixture of protease and phosphatase inhibitors
(Roche Diagnostics, Basel, Switzerland). The total collagen volume
in the liver was measured using a Sircol collagen assay
kit® (Biocolor Ltd., Carrickfergus, Northern Ireland).
The TGF-β levels in the liver were measured using ELISA (R&D
Systems, Minneapolis, MN, USA).
Determination of rat intestinal
permeability
A total of 25 mg fluorescein isothiocyanate
(FITC)-dextran (40 kDa; Sigma-Aldrich) was orally administered per
animal on the day of sacrifice. At 4 h after oral gavage of
FITC-dextran, each rat was anesthetized and blood was drawn from
its portal vein. The plasma was analyzed by fluorescence
measurement at an excitation wavelength of 490 nm and an emission
wavelength of 520 nm.
Statistical analysis
The results are presented as the mean ± standard
deviation and were analyzed using Student’s t-test for unpaired
data (SPSS version 22; IBM, Armonk, NY, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
General findings
The general findings of each experimental group at
the time of sacrifice are shown in Table I. The relative weights of the liver
in the CDAA group and the CDAA+AB group were significantly higher
than that of the CSAA group, whereas no significant differences
were observed between the CDAA and the CDAA+AB groups. Regarding
the serological data between the CDAA group and the CDAA+AB group,
no significant differences were observed in the levels of aspartate
aminotransferase, alanine aminotransferase, albumin, total
bilirubin, glucose, triglyceride, total cholesterol or high-density
lipoprotein cholesterol.
 | Table ICharacteristic features of the
experimental groups. |
Table I
Characteristic features of the
experimental groups.
| Characteristics | CSAA (n=5) | CDAA (n=10) | CDAA+AB (n=10) |
|---|
| Body weight (g) | 304.0±11.6 | 291.3±18.5 | 250.6±12.4a |
| Liver weight
(g) | 10.4±0.7 | 18.6±1.4a | 15.6±1.5a |
| Liver weight (%
body) | 3.4±0.2 | 6.4±0.2a | 6.2±0.4a |
| Aspartate
aminotransferase (IU/l) | 57.6±6.0 | 361.2±39.0a | 384.5±46.3a |
| Alanine
aminotransferase (IU/l) | 25.4±8.6 | 244.8±55.2a | 259.4±53.0a |
| Total bilirubin
(mg/dl) | 0.03±0.01 | 0.13±0.02a | 0.13±0.01a |
| Albumin (g/dl) | 3.0±0.2 | 3.3±0.3 | 3.1±0.2 |
| Total C
(mg/dl) | 42.6±5.9 | 26.1±3.3a | 24.6±2.1a |
| High density
lipoprotein C (mg/dl) | 13.4±3.2 | 14.3±2.5 | 14.9±1.6 |
| Triglyceride
(mg/dl) | 116.6±18.9 | 10.6±7.6a | 6.4±1.6a |
| Glucose
(mg/dl) | 135.0±28.8 | 101.9±11.3 | 105.3±29.6 |
Effect of poorly absorbable antibiotics
on liver fibrosis development
The present study initially examined the effects of
poorly absorbable antibiotics on liver fibrosis, induced by CDAA
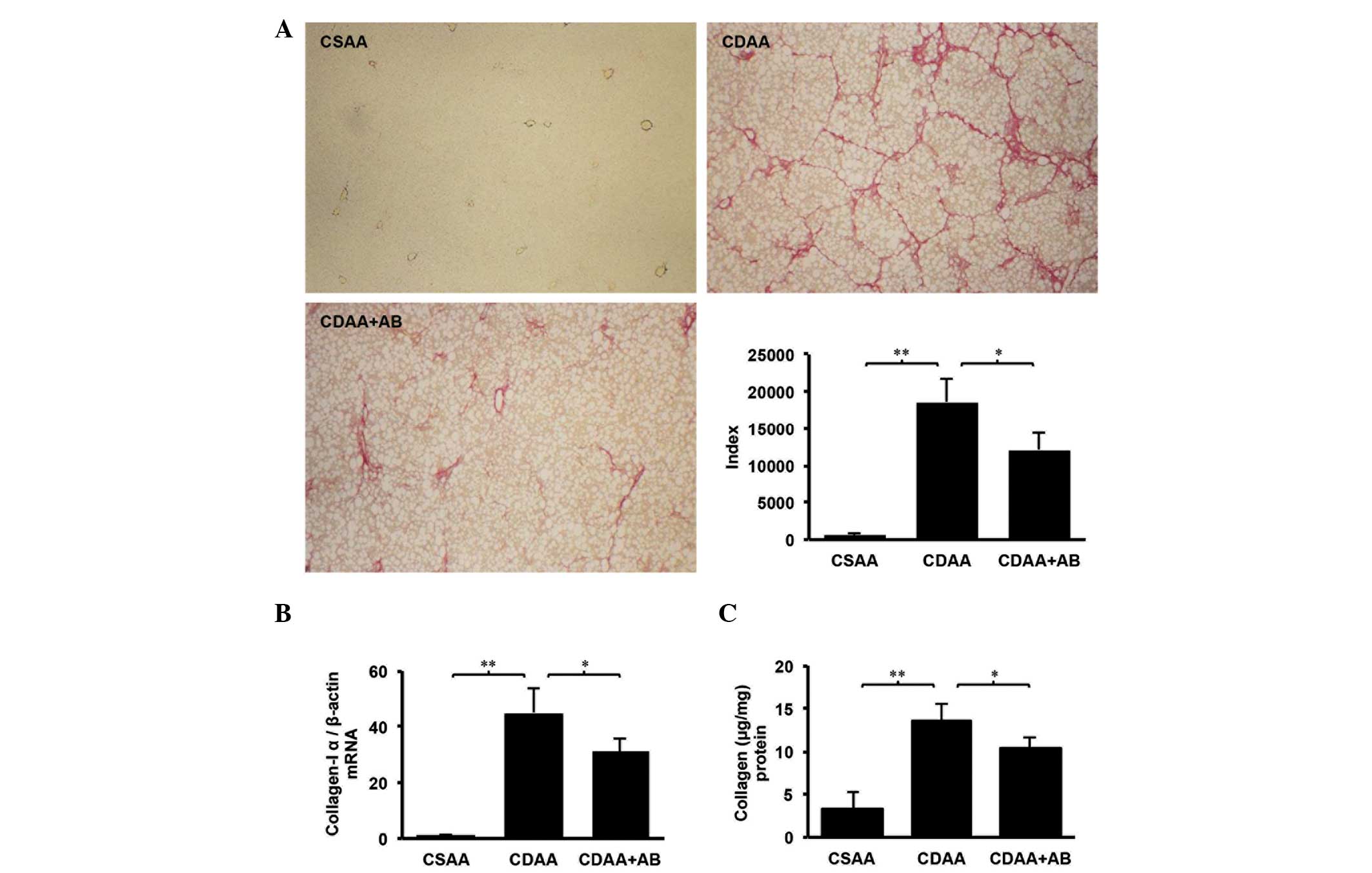
intake. As shown in Fig. 1A,
although marked fibrosis was observed in the CDAA group, no
fibrosis was identified in the CSAA control group and poorly
absorbable antibiotics attenuated the CDAA-induced fibrosis. Since
it is generally accepted that activated HSCs are critical in
fibrogenesis, immunohistochemical analysis of α-SMA was performed
to examine the effects of poorly absorbable antibiotics on HSC
activation during the development of liver fibrosis. Markedly
decreased levels of α-SMA expression were observed in the CDAA+AB
group (Fig. 2). Semi-quantitative
analysis performed using Image J software version 64 (National
Institutes of Health, Bethesda, MD, USA) revealed a significant
decrease of α-SMA in the CDAA+AB group compared with the CDAA group
(Fig. 2A). Additionally, markedly
suppressed levels of hepatic TGF-β and total collagen were revealed
in the CDAA+AB group, compared with the CDAA group (Figs. 1C and 2C). RT-qPCR also revealed that these
inhibitory effects were closely correlated with alterations in mRNA
expression levels of TGF-β and collagen-Iα (Figs. 1B and 2B). The results of the present study
suggested that poorly absorbable antibiotics attenuated HSC
activation and liver fibrosis via control of TGF-β and collagen in
the experimental hepatic fibrosis model.
Effect of poorly absorbable antibiotics
on LPS-TLR4 signaling
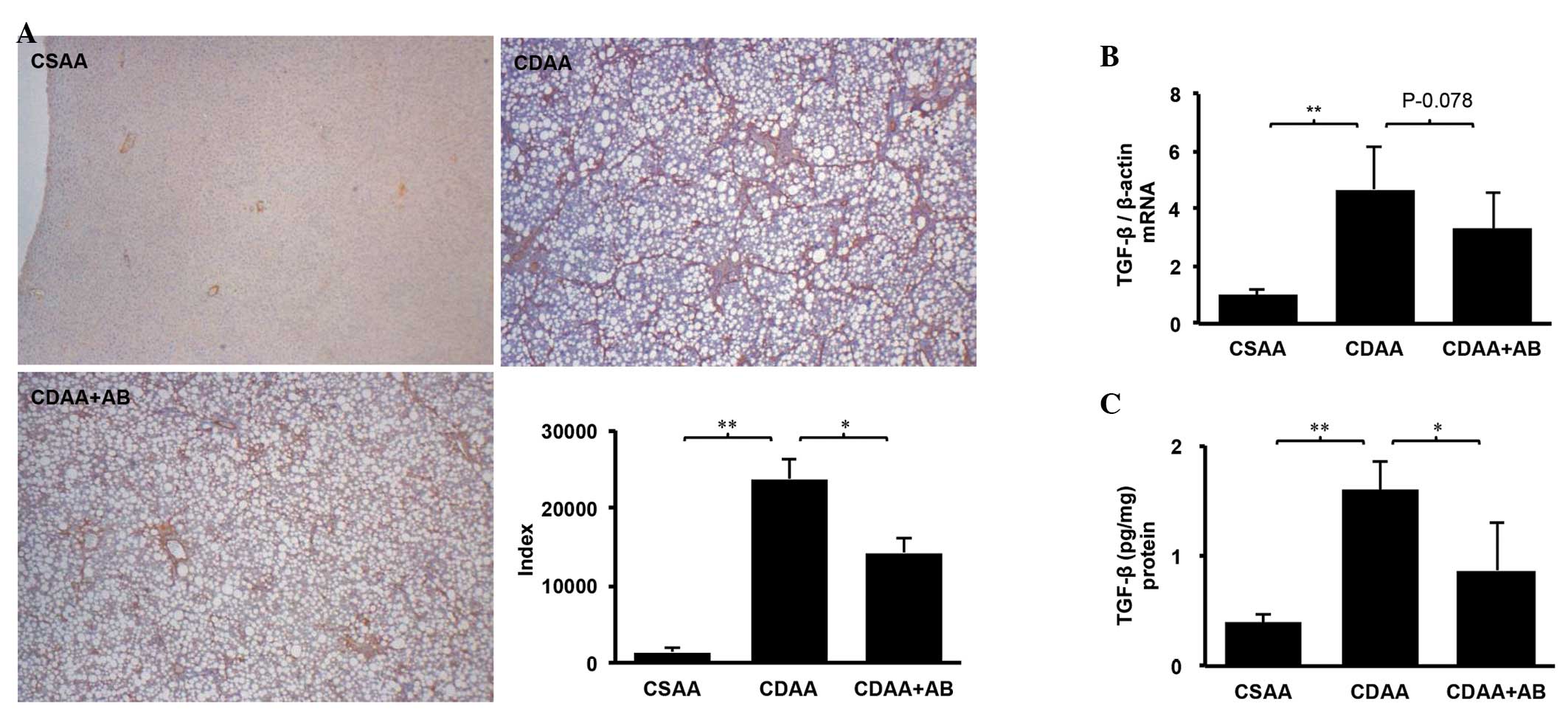
TLR4 enhances hepatic inflammation and fibrogenesis
(8,18). This finding led to the hypothesis
of the present study that poorly absorbable antibiotics may
attenuate LPS-TLR4 signaling, with liver fibrosis ameliorated as a
result. TLR4 mRNA expression in the liver and intestine were then
examined. Notably, TLR4 mRNA level in the liver was elevated in the
CDAA group and the CDAA-induced increase was significantly
decreased by antibiotics (Fig.
3A). However, TLR4 mRNA levels in the intestine were similar in
all groups (Fig. 3B). These data
suggested that TLR4-associated signaling in the intestine was not
important for liver fibrosis, whereas TLR4 in the liver was
essential for liver fibrosis. Subsequently, the mRNA levels of LBP
were measured, which is essential for LPS to bind TLR4 and is
correlated with serum endotoxin levels (24). A significantly elevated mRNA level
of LBP was observed in the CDAA group and this increase was reduced
in the CDAA+AB group (Fig.
3C).
Effect of poorly absorbable antibiotics
on intestinal permeability and tight junction protein (TJP)
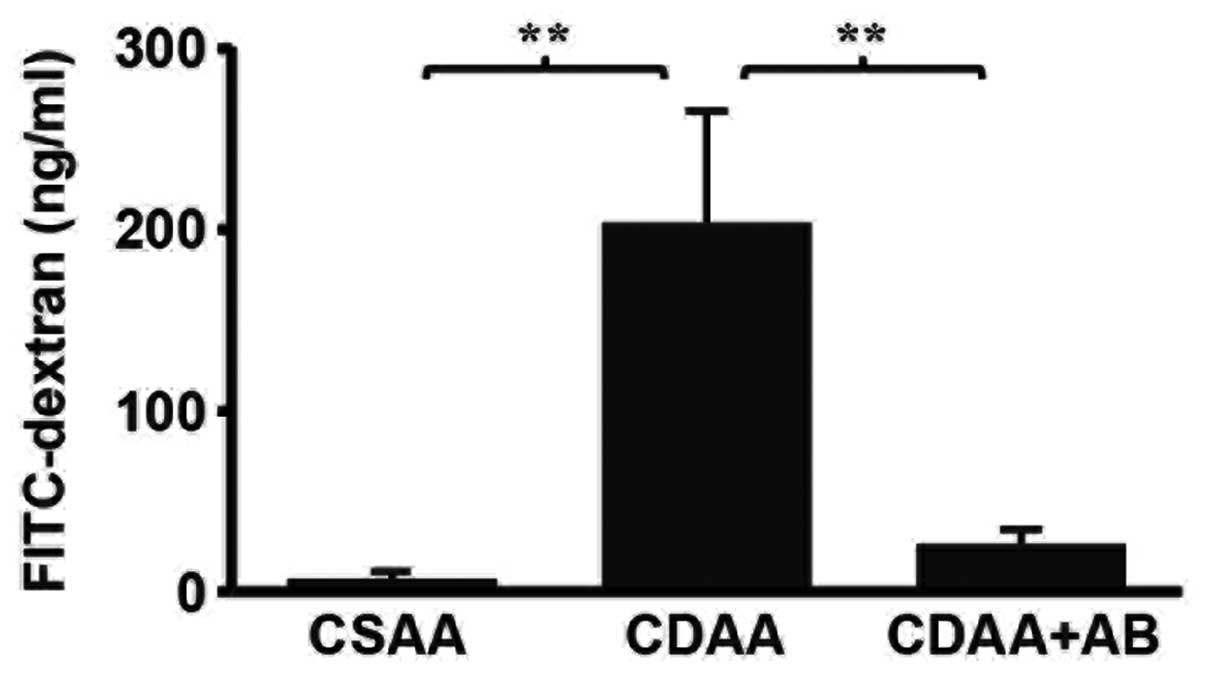
The elevated mRNA level of LBP suggested that the
serum LPS level was increased in the CDAA group. The serum LPS
level was hypothesized to be involved in gut permeability,
therefore gut permeability was examined by analyzing the
fluorescence levels of the portal vein following oral gavage
loading with FITC-dextran. The fluorescence levels of the portal
vein in the CDAA group were increased when compared with the CSAA
group. The increase of intestinal permeability of the CDAA group
was improved by the addition of poorly absorbable antibiotics
(Fig. 4). Since gut permeability
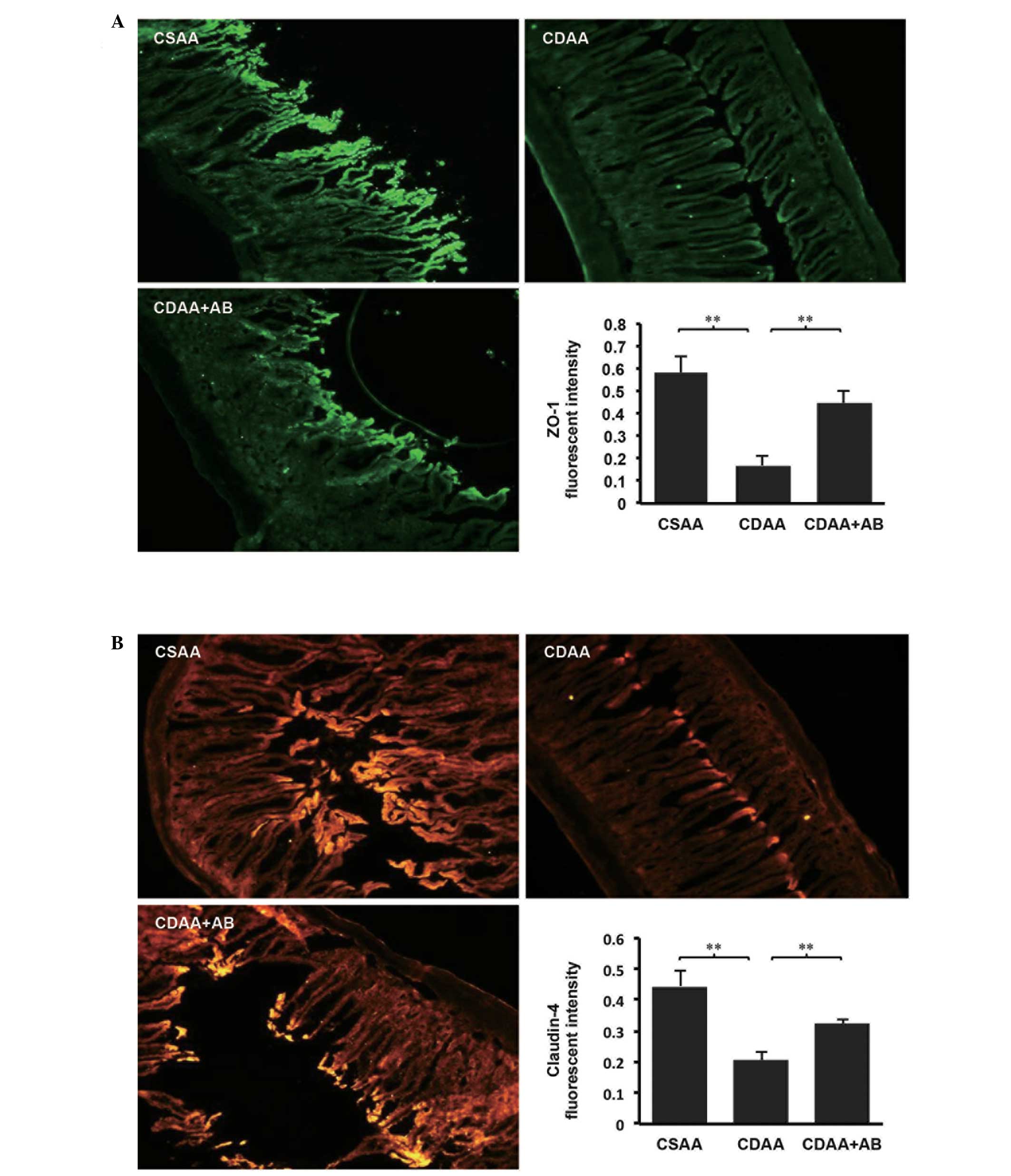
is controlled by TJPs, including ZO-1 and Claudin-4 (14,20),
the immunohistochemical analyses of ZO-1 and Claudin-4 in the
intestinal sections were examined. As shown in Fig. 5, immunohistochemical analyses
revealed that significant expression levels of ZO-1 and Claudin-4
were predominant in the intestinal sections of the CSAA control
group (Fig. 5A and B). By
contrast, the delocalization and substantial decrease in the
intestinal sections of the CDAA group were significantly improved
by poorly absorbable antibiotic administration.
Discussion
In the present study, the effect of the poorly
absorbable antibiotics, polymyxin and neomycin, on the development
of hepatic fibrosis and intestinal permeability was examined. It
was demonstrated that the antibiotics not only reduced CDAA-induced
hepatic fibrosis and HSC activation but also improved intestinal
permeability.
The liver is the main target of intestinally-derived
bacterial products and the rate of bacterial translocation
increases in various models of hepatic disease, rendering LPS a
possible candidate mediator of TLR4-dependent profibrogenic
effects. Accordingly, increased LBP mRNA expression was identified
in the CDAA group, indicating that LPS was increased. In addition,
LBP mRNA expression and fibrogenesis were reduced in rats treated
with poorly absorbable antibiotics, suggesting that the intestinal
flora is the main source of LPS and that intestinally-derived LPS
drives fibrogenesis.
Translocated LPS derived from the gut microflora is
considered to mediate TLR4 activation in the liver. However, this
translocation may be independent of intestinal TLR4 (21). The mRNA expression levels of TLR4
in the liver and intestine were assessed in the present study. The
mRNA expression levels in the liver of the CDAA-induced NASH model
were increased. By contrast, the mRNA levels in the intestine of
the CDAA-induced NASH model were not increased. However, Guo et
al (22) reported that LPS
caused an increase in intestinal permeability via an intracellular
mechanism involving the TLR4-dependent upregulation of CD14
membrane expression. The association between LPS and TLR4 in
intestinal permeability remains controversial.
NAFLD is associated with increased intestinal
permeability and small intestinal bacteria overgrowth (21,23).
These findings have been considered to be associated with the
severity of hepatic steatosis. Increased intestinal permeability
may be a condition supporting the hypothesis of the contribution of
the gut-liver axis to the development of NAFLD (14). The intestinal barrier defect may be
caused by disruption of the tight junction proteins between
intestinal epithelial cells, allowing substances, including
lipopolysaccharides, to pass from the intestine to the portal vein,
imbalance of proliferation and apoptosis, intestinal mucosal
atrophy and edema, which is associated with portal hypertension or
the absence of bile acids and systemic increases in inflammatory
cytokines and oxidative stress produced in the liver (24–26).
LPS causes an increase in intestinal permeability via an
intracellular mechanism involving the TLR4-dependent upregulation
of CD14 membrane expression (22).
Caco-2 cells grown in zinc-deficient media have
reduced transepithelial electrical resistance and altered
expression levels of ZO-1 and occludin, which are intestinal tight
junction proteins, compared with Caco-2 cells grown in zinc-replete
media (27). In clinical practice,
zinc deficiency is common in patients with liver cirrhosis
(28,29). In in vitro study, Caco-2
cells, which mimic intestinal epithelial cells, grown in
zinc-deficient media have reduced TJP; therefore it was
hypothesized that zinc deficiency may potentially be relevant to
the increased intestinal permeability. In the NASH model used in
the present study, CDAA-induced hepatic fibrosis, endogenous LPS
and systemic increases in inflammatory cytokines may disrupt
intestinal tight junction proteins. Considering these findings, the
recruitment of TJPs using probiotics and zinc preparations, for
example, offers a novel strategy for NASH treatment.
The intestinal microflora is involved in liver
fibrosis. In the present in vivo model, dietary habits,
which increase the percentage of intestinal endotoxin producers,
including Gram-negative bacteria may accelerate liver fibrogenesis,
introducing dysbiosis as a cofactor contributing to chronic liver
injury in NAFLD (30). Endo et
al (9) also demonstrated that
butyrate-producing probiotics reduced NAFLD progression in rats.
These data indicated that intestinal microflora may be a new target
for NASH treatment.
In conclusion, the inhibition of LPS-TLR4 signaling
with poorly absorbable antibiotics attenuated the liver fibrosis
development in NASH via inhibition of HSC activation. These results
indicated that reduction of LPS and restoration of the intestinal
TJPs may be a new therapeutic strategy for the treatment of the
development of liver fibrosis in NASH.
Abbreviations:
|
TLR4
|
toll-like receptor 4
|
|
NASH
|
non-alcoholic steatohepatitis
|
|
LPS
|
lipopolysaccharide
|
|
CDAA
|
choline deficiency amino acid
|
|
HSC
|
hepatic stellate cell
|
|
LBP
|
LPS binding protein
|
|
CSAA
|
choline supplemented amino acid
|
|
TJP
|
tight junction protein
|
|
NAFLD
|
non-alcoholic fatty liver disease
|
|
HCC
|
hepatocellular carcinoma
|
|
TGF-β
|
transforming growth factor-β
|
|
α-SMA
|
α-smooth muscle actin
|
|
Glu
|
glucose
|
|
TG
|
triglyceridel
|
References
|
1
|
Angulo P: Nonalcoholic fatty liver
disease. N Engl J Med. 346:1221–1231. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Henao-Mejia J, Elinav E, Jin C, et al:
Inflammasome-mediated dysbiosis regulates progression of NAFLD and
obesity. Nature. 482:179–185. 2012.PubMed/NCBI
|
|
3
|
Ekstedt M, Franzen LE, Mathiesen UL, et
al: Long-term follow-up of patients with NAFLD and elevated liver
enzymes. Hepatology. 44:865–873. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
El-Serag HB: Hepatocellular carcinoma. N
Engl J Med. 365:1118–1127. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rivera CA, Adegboyega P, van Rooijen N,
Tagalicud A, Allman M and Wallace M: Toll-like receptor-4 signaling
and Kupffer cells play pivotal roles in the pathogenesis of
non-alcoholic steatohepatitis. J Hepatol. 47:571–579. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Akira S and Takeda K: Toll-like receptor
signaling. Nat Rev Immunol. 4:499–511. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jagavelu K, Routray C, Shergill U, O’Hara
SP, Faubion W and Shah VH: Endothelial cell toll-like receptor 4
regulates fibrosis-associated angiogenesis in the liver.
Hepatology. 52:590–601. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Seki E, de Minicis S, Osterreicher CH, et
al: TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med.
13:1324–1332. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Endo H, Niioka M, Kobayashi N, Tanaka M
and Watanabe T: Butyrate-producing probiotics reduce nonalcoholic
fatty liver disease progression in rats: New insight into the
probiotics for the gut-liver axis. PLoS One. 8:e633882013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ruiz AG, Casafont F, Crespo J, et al:
Lipopolysaccharide-binding protein plasma levels and liver
TNF-alpha gene expression in obese patients: Evidence for the
potential role of endotoxin in the pathogenesis of non-alcoholic
steatohepatitis. Obes Surg. 17:1374–1380. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Musso G, Gambino R and Cassader M: Gut
microbiota as a regulator of energy homeostasis and ectopic fat
deposition: mechanisms and implications for metabolic disorders.
Curr Opin Lipidol. 21:76–83. 2010. View Article : Google Scholar
|
|
12
|
Szabo G, Bala S, Petrasek J and Gattu A:
Gut-liver axis and sensing microbes. Dig Dis. 28:737–744. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nguyen AT, Mandard S, Dray C, et al:
Lipopolysacch arides-mediated increase in glucose-stimulated
insulin secretion: involvement of the GLP-1 pathway. Diabetes.
63:471–482. 2014. View Article : Google Scholar
|
|
14
|
Miele L, Valenza V, La Torre G, et al:
Increased intestinal permeability and tight junction alterations in
nonalcoholic fatty liver disease. Hepatology. 49:1877–1887. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ruberto F, Ianni S, Babetto C, et al:
Polymyxin-B endotoxin removal device: making the point on
mechanisms of action, clinical effectiveness and possible future
applications: review. Infect Disord Drug Targets. 13:128–32. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yoshiji H, Kuriyama S, Yoshii J, et al:
Angiotensin-II type 1 receptor interaction is a major regulator for
liver fibrosis development in rats. Hepatology. 34:745–750. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kaji K, Yoshiji H, Ikenaka Y, et al:
Dipeptidyl peptidase-4 inhibitor attenuates hepatic fibrosis via
suppression of activated hepatic stellate cell in rats. J
Gastroenterol. 49:481–491. 2014. View Article : Google Scholar
|
|
18
|
Roh YS and Seki E: Toll-like receptors in
alcoholic liver disease, non-alcoholic steatohepatitis and
carcinogenesis. J Gastroenterol Hepatol. 28:38–42. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schumann RR: Old and new findings on
lipopolysaccharide-binding protein: a soluble pattern-recognition
molecule. Biochem Soc Trans. 39:989–993. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ulluwishewa D, Anderson RC, McNabb WC,
Moughan PJ, Wells JM and Roy NC: Regulation of tight junction
permeability by intestinal bacteria and dietary components. J Nutr.
141:769–776. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Seki E and Schnabl B: Role of innate
immunity and the microbiota in liver fibrosis: crosstalk between
the liver and gut. J Physiol. 590:447–458. 2012. View Article : Google Scholar :
|
|
22
|
Guo S, Al-Sadi R, Said HM and Ma TY:
Lipopolysaccharide causes an increase in intestinal tight junction
permeability in vitro and in vivo by inducing enterocyte membrane
expression and localization of TLR-4 and CD14. Am J Pathol.
182:375–387. 2013. View Article : Google Scholar :
|
|
23
|
Brun P, Castagliuolo I, Di Leo V, et al:
Increased intestinal permeability in obese mice: new evidence in
the pathogenesis of nonalcoholic steatohepatitis. Am J Physiol
Gastrointest Liver Physiol. 292:G518–G525. 2007. View Article : Google Scholar
|
|
24
|
Du Plessis J, Vanheel H, Janssen CE, et
al: Activated intestinal macrophages in patients with cirrhosis
release NO and IL-6 that may disrupt intestinal barrier function. J
Hepatol. 58:1125–1132. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Assimakopoulos SF, Tsamandas AC,
Tsiaoussis GI, et al: Intestinal mucosal proliferation, apoptosis
and oxidative stress in patients with liver cirrhosis. Ann Hepatol.
12:301–307. 2013.PubMed/NCBI
|
|
26
|
Assimakopoulos SF, Tsamandas AC, Louvros
E, et al: Intestinal epithelial cell proliferation, apoptosis and
expression of tight junction proteins in patients with obstructive
jaundice. Eur J Clin Invest. 41:117–125. 2011. View Article : Google Scholar
|
|
27
|
Finamore A, Massimi M, Conti Devirgiliis L
and Mengheri E: Zinc deficiency induces membrane barrier damage and
increases neutrophil transmigration in Caco-2 cells. J Nutr.
138:1664–1670. 2008.PubMed/NCBI
|
|
28
|
Chiba M, Katayama K, Takeda R, et al:
Diuretics aggravate zinc deficiency in patients with liver
cirrhosis by increasing zinc excretion in urine. Hepatology
Research. 43:365–373. 2013. View Article : Google Scholar
|
|
29
|
Mohammad MK, Zhou Z, Cave M, Barve A and
McClain CJ: Zinc and liver disease. Nutr Clin Pract. 27:8–20. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
De Minicis S, Rychlicki C, Agostinelli L,
et al: Dysbiosis contributes to fibrogenesis in the course of
chronic liver injury in mice. Hepatology. 59:1738–1749. 2014.
View Article : Google Scholar
|