Introduction
Fibroblast growth factor 9 (FGF9) is important in a
variety of biological processes, including the development of the
lung, limb, testis and skeletal system in vertebrates (1–7). In
skeletal development, FGF9 may have complex and important roles in
endochondral ossification and intramembranous bone formation
(4,5,7).
Within the context of long bone development, Hung et al
(4) demonstrated that FGF9
promotes chondrocyte hypertrophy in the early stages of skeletal
development and regulated the vascularization of the growth plate
and osteogenesis in the later stages. In addition, mice lacking
FGF9 exhibited decreased chondrocyte proliferation and delayed
onset of chondrocyte hypertrophy defects in skeletal
vascularization, which led to abnormal osteogenesis. Behr et
al (7) revealed that FGF9
promoted bone healing by initiating angiogenesis through vascular
endothelial growth factor (VEGF)-α, which further supported the
concept that the healing processes observed in adults may be a
recapitulation of those observed during embryonic skeletal
development. Ignelzi et al (8) demonstrated that exogenous FGF9 leads
to increased expression of Msh homeobox 2, followed by osteogenic
differentiation induced by the sutural mesenchyme during
craniofacial skeletal development in mouse calvaria. Furthermore,
Govindarajan and Overbeek (5)
revealed that the differentiation of cranial mesenchymal cells from
the mesoderm, but not cranial neural crest cells, can be altered by
FGF9 from intramembranous to endochondral ossification. Fakhry
et al (9) also demonstrated
that in cell populations comprising mature osteoblasts,
proliferation is stimulated by FGF9; however, this does not occur
in populations of undifferentiated precursor cells. Although these
findings collectively suggested that FGF9 is able to promote bone
formation, other studies have demonstrated contradictory results.
Weksler et al (10)
indicated that FGF9 weakly promoted the proliferation of a rat
calvaria-derived cell line in vitro, but inhibited its
terminal differentiation. Garofalo et al (11) observed that, in transgenic mice,
the proliferation and terminal differentiation of chondrocytes in
the cartilage was inhibited following induced overexpression of
FGF9. In addition, FGF9-FGFR3 signaling physiologically inhibited
endochondral ossification, leading to a phenotype which is
characteristic of skeletal dysplasia (11). Wu et al (12) demonstrated that a loss-of-function
mutation in FGF9 led to the proliferation and differentiation of
chondrocytes, with an increase in osteogenic differentiation and
matrix mineralization in bone marrow-derived mesenchymal stem cells
(BMSCs) and joint ankylosis. Additionally, a previous study by our
group revealed that, in dental pulp stem cells (DPSCs), exogenous
FGF9 led to the promotion of chondrogenesis and partial inhibition
of mineralization (13).
Although previous studies have revealed that FGF9 is
important in skeletal development, their apparently contradictory
results also indicated that the action of FGF9 is complex and that
the biological effects of FGF9 may depend on the gene dosage,
developmental stage, cell type and interactions with other
cytokines. In addition, the activities of genes downstream of FGF9,
including FGF receptor 3 and mitogen-activated protein-kinase
(MAPK), which include extracellular signal-regulated kinase (ERK),
p38 and c-Jun N-terminal kinase (JNK), may also affect its function
(12–14). However, the different types of MAPK
are activated by different extracellular stimuli and differ in
their downstream targets, therefore having distinct roles in
cellular responses. In the present study, the effects of FGF9 on
osteogenic differentiation in BMSCs and DPSCs were compared to
investigate whether FGF9 differs in its osteogenic induction
capabilities in different cells. In addition, the present study
examined whether ERK1/2, a gene downstream of FGF9, differs in its
role in osteogenesis in different types of cell.
Materials and methods
Culture of BMSCs and DPSCs
The method of the present study was reviewed and
approved by the Ethical Committee of Shanghai Ninth People’s
Hospital, Shanghai Jiao Tong University School of Medicine
(Shanghai, China). DPSCs were isolated using direct cell outgrowth
from pulp tissue explants and cultured as previously reported
(13). Briefly, healthy human
third molars were collected from adults aged between 16 and 25
years (mean, 18±3.2) at the Oral and Maxillary Facial Surgery
Clinic of The Ninth People’s Hospital, Shanghai Jiao Tong
University School of Medicine. All individuals provided informed
consent. Following cleaning of the teeth, the pulp chambers were
accessed by cutting the cementoenamel junction with a sterile
fissure dental bur. Following exposure of the pulp, the pulp tissue
was removed in fragments of ~0.5 mm, which were then placed onto a
6-cm culture dish containing Dulbecco’s modified Eagle’s medium
(DMEM; Gibco-BRL, Grand Island, NY, USA) supplemented with 20%
fetal bovine serum (FBS; Hyclone, Logan, UT, USA) and antibiotics
(Gibco-BRL) prior to incubation at 37°C in 5% CO2. At
confluence, the outgrown cells were transferred onto a 10-cm dish
and then continuously passaged for further experiments. All
experiments were performed with mixed cells from multiple patients
and cells from the third passage were used.
Four-week-old male Sprague Dawley rats were obtained
from the Ninth People’s Hospital Animal Center (Shanghai, China)
and the bone marrow was extracted from the femurs and tibias, as
described previously (15). All
procedures were approved by the Animal Research Committee of the
Shanghai Ninth People’s Hospital. The BMSCs were cultured in DMEM
supplemented with 10% FBS and 1% penicillin/streptomycin at 37°C in
5% CO2 for 5–7 days. When they had reached 80–90%
confluence, the BMSCs were transferred onto a 10-cm dish and then
continuously passaged for further experiments. The culture medium
was replaced every three days and the BMSCs at passages two to four
were used in subsequent experiments.
Osteogenic induction (OI)
The osteogenic medium used for the differentiation
of the cultured BMSCs and DPSCs comprised DMEM, 10% FBS,
10−8 M dexamethasone, 5 mmol/l
KH2PO4, 50 mg/ml L-ascorbic acid and 50 mg/ml
gentamycin (Sigma-Aldrich, St. Louis, MO, USA). The medium was
replaced every 2–3 days for 14–28 days. The conditions used in the
different experiments were as follows: BMSCs/DPSCs cultured in DMEM
with 10% FBS (control; CON group); BMSCs/DPSCs cultured in the OI
medium described above (OI group); BMSCs/DPSCs cultured in OI
medium with 20 ng/ml FGF9 protein (OI+FGF9 group) and
BMSCs/DPSCs/CSCs cultured in OI medium with 20 ng/ml FGF9 protein,
U0126, SB203580 and SP600125 (OI+FGF9+U/SB/SP group). The
inhibitors U0126, SB203580 and SP600125 at concentrations of 10, 10
and 25 μM, respectively, were added to the DMEM medium 2 h prior to
its replacement with OI medium, which also contained these three
inhibitors, for continuous culture.
MTT assay
A total of 104 BMSCs/well were seeded
into 96-well plates and subsequently cultured in 200 μl DMEM with
10% FBS and exogenous FGF9 protein at different concentrations (1,
5, 10, 20 or 40 ng/ml). The proliferation of the BMSCs was then
evaluated at different time-points (1, 3, 5, 7 and 9 days) using an
MTT assay. Briefly, the BMSCs were incubated with 0.5 mg/ml MTT
(Sigma-Aldrich) under normal culture conditions for another 4 h.
The medium was then removed, 200 μl DMSO (Sigma-Aldrich) was added
to each well and the plates were agitated for 10 min. The
absorbance of the solution was then measured at 490 nm and
corrected with an NP blank using a spectrophotometer (Elx 800;
BioTek Instruments, Inc., Winooski, VT, USA). A growth curve of the
BMSCs was then generated based on the absorbance values measured at
different time-points.
Alkaline phosphatase (ALP) staining and
alizarin red staining
ALP staining of the BMSCs and DPSCs in vitro
was performed 7 days after treatment using an ALP staining kit
(Biyuntian Biotech Co., Ltd., Shanghai, China). Briefly, the
cultured cells were washed twice using phosphate-buffered saline
(PBS; Gibco-BRL), fixed in 4% paraformaldehyde and stained using an
ALP staining kit for 30 min. This was followed by thorough washing
and images were then captured with a digital single lens reflex
camera (Olympus, Osaka, Japan). For the alizarin red S staining,
the cultured cells were fixed in 95% ethanol 21 days after
treatment. The cells were then stained using 1% Alizarin Red S
(Sigma-Aldrich) for 20 min. Subsequently, the cell samples were
thoroughly washed and images were captured.
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR)
Total cellular RNA was extracted from different cell
samples using TRIzol reagent (Invitrogen Life Technologies,
Carlsbad, CA, USA) according to the manufacturer’s instructions.
The total RNA was converted to cDNA using a Prime Script-RT reagent
kit (Takara Bio Inc., Shiga, Japan) according to the manufacturer’s
instructions. The gene-specific primers used for the amplification
of collagen type 1 (COL1), runt-related transcription factor 2
(Runx2), osteopontin (OPN) and osteocalcin (OCN) are listed in
Table I. All RT-qPCR reactions
were performed using a SYBR Green system according to the
manufacturer’s instructions with 20 μl reaction volumes in 96-well
microwell plates and using a MyiQ Real-Time PCR Detection system
(Bio-Rad, Hercules, CA, USA) (13). Data analysis was based on
calculating the relative expression levels of these genes compared
with the controls.
 | Table IPrimer sequences for reverse
transcription quantitative polymerase chain reaction. |
Table I
Primer sequences for reverse
transcription quantitative polymerase chain reaction.
| Gene name | Primer sequence |
|---|
| Rat GAPDH | F,
5′-CCTGCACCACCAACTGCTTA-3′
R, 5′-GGCCATCCACAGTCTTCTGAG-3′ |
| Rat Runx2 | F,
5′-TCTCTGACCGCCTCAGTGATT-3′
R, 5′-TGTGTCTGCCTGGGATCTGTA-3′ |
| Rat COL1 | F,
5′-CTGCCCAGAAGAATATGTATCACC-3′
R, 5′-GAAGCAAAGTTTCCTCCAAGACC-3′ |
| Rat OPN | F,
5′-GGAGTCCGATGAGGCTATCAA-3′
R, 5′-TCCGACTGCTCAGTGCTCTC-3′ |
| Rat OCN | F,
5′-GCCCTGACTGCATTCTGCCTCT-3′
R, 5′-TCACCACCTTACTGCCCTCCTG-3′ |
| Human GAPDH | F,
5′-TTCGACAGTCAGCCGCATCTT-3′
R, 5′-ATCCGTTGACTCCGACCTTCA-3′ |
| Human Runx2 | F,
5′-GCCTTCAAGGTGGTAGCCC-3′
R, 5′-CGTTACCCGCTATGACAGTA-3′ |
| Human COL1 | F,
5′-TCCAACGAGATCGAGATCC-3′
R, 5′-AAGCCGAATTCCTGGTCT-3′ |
| Human OPN | F,
5′-CATGAGAATTGCAGTGTTTGCT-3′
R, 5′-CTTGCAAGGGTCTGTGGGG-3′ |
| Human OCN | F,
5′-CCCCCTCTAGCCTAGGACC-3′
R, 5′-ACCAGGTAATGCCAGTTTGC-3′ |
Western blot analysis
The BMSCs and DPSCs were collected and washed three
times using ice-cold PBS. The samples were then lysed using
radioimmunoprecipitation assay buffer containing protease
inhibitors (Biyuntian Biotech Co., Ltd.). Subsequently, the lysates
were centrifuged and the supernatants were boiled in SDS sample
buffer (Sigma-Aldrich). The samples were then separated by 10%
SDS-PAGE and transferred onto a polyvinylidene difluoride membrane
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). The membranes were
inhibited using 5% milk for 2 h and then incubated with rabbit
anti-mouse ERK1/2, p-ERK1/2, p38, phospho (p)-p38 or tubulin
antibodies (1:1,000 dilutions; Cell Signaling Technology Inc.,
Danvers, MA, USA) or goat anti-mouse Runx2 or COL1 antibodies
(1:1,000; R&D Systems, Minneapolis, MN, USA) in PBS with
Tween-20 buffer overnight. Finally, the membranes were incubated
with horseradish peroxidase-conjugated secondary antibody (1:3,000,
Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) and visualized
using an enhanced chemiluminescence western blotting kit (Pierce
Biotechnology, Inc., Rockford, IL, USA).
Statistical analysis
The data are expressed as the mean ± standard
deviation. Data analysis was performed using an independent samples
test with SPSS 18.0 software (International Business Machines,
Armonk, NY, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Effect of exogenous FGF9 on the
proliferation of BMSCs in vitro
The effects of exogenous FGF9 on BMSC viability and
proliferation were examined using an MTT assay, with BMSCs cultured
in regular DMEM medium as a control. The FGF9-treated cells
exhibited a several-fold increase in cell number during the first
three days of culture. Subsequently, the control cells demonstrated
a slight increase in cell number, whereas a substantial increase in
cell number was observed in the cells treated with FGF9 between
days three and nine. However, no significant differences were
observed in proliferation among the cells treated with exogenous
FGF9 at different concentrations (1, 5, 10, 20 and 40 ng/ml;
Fig. 1). Therefore, the median
concentration of 20 ng/ml was used in the subsequent experiments
(Fig. 1).
 | Figure 1Effect of exogenous FGF9 on BMSC
proliferation. The effects of different concentrations of exogenous
FGF9 on the proliferation of BMSCs were examined using an MTT assay
on days 1, 3, 5, 7 and 9. BMSCs were cultured in Dulbecco’s
modified Eagle’s medium as a control. The FGF9-treated cells
exhibited a several-fold increase in cell proliferation during the
first three days of culture. Statistically significant differences
were detected between the control and FGF9-treated cells, whereas
no statistically significant differences were detected among the
treated cells with different concentrations of FGF9 (1, 5, 10, 20
and 40 ng/ml). Data are expressed as the mean ± standard deviation
of three independent experiments. BMSC, bone marrow stromal stem
cell; Con, control; FGF9, fibroblast growth factor 9. |
Effect of exogenous FGF9 on the
osteogenic differentiation of BMSCs in vitro
ALP staining, RT-qPCR and western blot analysis were
performed to investigate the effect of exogenous FGF9 on the
osteogenic differentiation of BMSCs in vitro. As shown in
Fig. 2A, the ALP and alizarin red
staining demonstrated that FGF9 inhibited the osteogenic
differentiation of BMSCs in a concentration-dependent manner. The
RT-qPCR and western blot analyses revealed that the gene expression
levels of four osteogenic markers, Runx2, COL1, OPN and OCN, were
reduced by treatment with 20 ng/ml exogenous FGF9 for 12 h and 7,
14 and 21 days. This further confirmed that exogenous FGF9 impaired
the osteogenic capacity of BMSCs (Fig.
2B–F).
 | Figure 2Effect of exogenous FGF9 on the
osteogenic differentiation of BMSCs in vitro. (A) ALP
staining of cultured monolayer BMSCs at day 7 and alizarin red
staining at day 21. FGF9 inhibited the ALP expression and matrix
mineralization of BMSCs in a concentration-dependent manner. (B)
Effects of FGF-9 on Runx2 protein levels in BMSCs 12 h after
treatment were investigated by western blot analysis. (C–F) mRNA
expression levels of four osteogenic transcription factors, Runx2,
COL1, OPN and OCN, at different stages of osteogenesis in the
FGF9-treated (20 ng/ml) BMSCs were investigated using reverse
transcription quantitative polymerase chain reaction (12 h and 7,
14 and 21 days after treatment, respectively). The gene and protein
expression levels of these four osteogenic transcription factors
were reduced by exogenous FGF9. Data are expressed as the mean ±
standard deviation of three independent experiments.
*P<0.05, **P<0.01. BMSCs, bone marrow
stromal stem cells; ALP, alkaline phosphatase; Runx2, runt-related
transcription factor 2; COL1, collagen type 1; OPN, osteopontin;
OCN, osteocalcin; CON, Dulbecco’s modified Eagle’s medium; OI,
osteogenic induction medium; FGF9, fibroblast growth factor 9. |
Effect of exogenous FGF9 on the
phosphorylation of ERK1/2, p38 and JNK in BMSCs in vitro
Previous studies have revealed that the MAPK
signaling pathway is important in the osteogenic differentiation of
mesenchymal stem cells. The results of the western blot analysis in
the present study revealed that p38, ERK1/2 and JNK phosphorylation
were augmented in the BMSCs cultured in the osteogenic medium
compared with the controls (Fig.
3A). Furthermore, the incubation of BMSCs cultured in
osteogenic medium containing 20 ng/ml FGF9 significantly enhanced
MAPK signaling, leading to increased levels of phosphorylated
ERK1/2, p38 and JNK, without altering the total quantities of these
proteins. The increased levels of phosphorylated ERK1/2 were first
observed at 5 min and peaked 5 min after FGF9 treatment,
maintaining a high level thereafter. The level of phosphorylated
p38 also increased and peaked at 15 min prior to gradually
reducing. Similarly, phosphorylated JNK increased and peaked at 30
min (Fig. 3A).
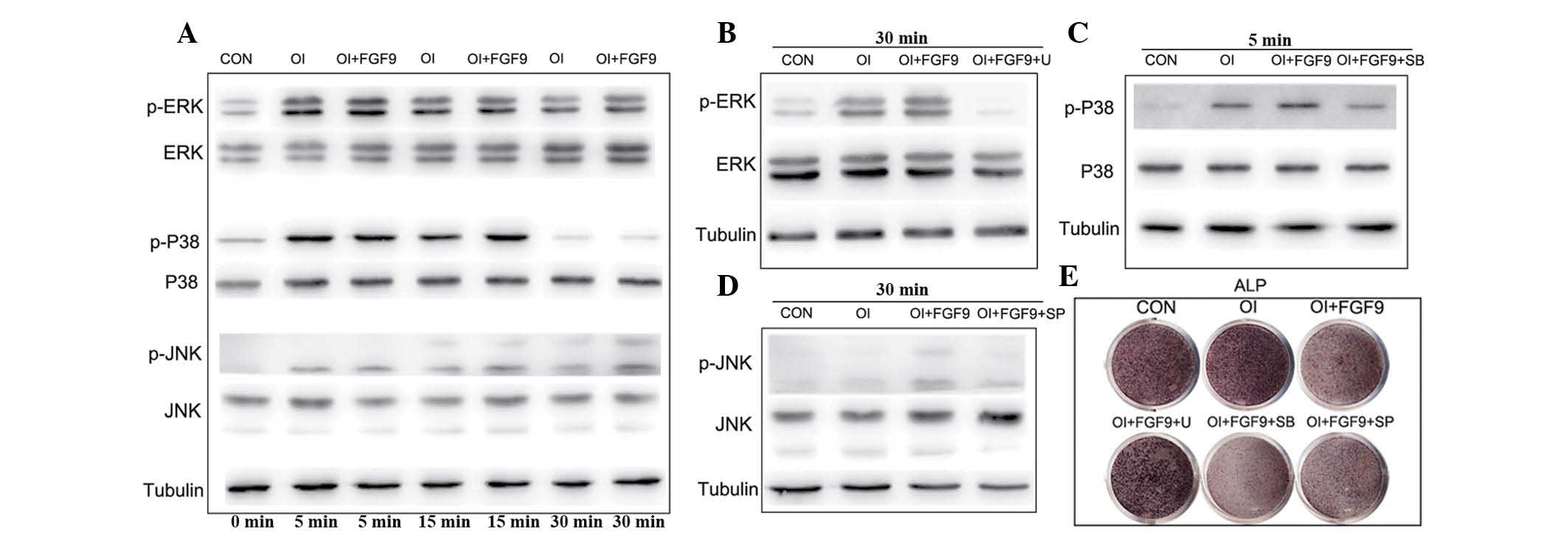 | Figure 3Effect of the FGF9/mitogen-activated
protein kinase signaling pathway in the osteogenic differentiation
of BMSCs in vitro. (A) ERK1/2, p-ERK1/2, p38, p-p38, JNK and
p-JNK protein levels in FGF9-treated (20 ng/ml) BMSCs were measured
by western blot analysis at the indicated time-points. Tubulin was
used to demonstrate equal loading of all samples. FGF9
significantly enhanced the phosphorylation of ERK1/2, P38 and JNK
in BMSCs compared with that in the CON and OI groups. (B–D)
Cultured BMSCs were pretreated with the protein synthesis
inhibitors, ERK1/2 inhibitor U0126 (10 μM), p38 inhibitor SB203580
(10 μM) and JNK inhibitor SP600125 (25 μM), 2 h prior to
experiments. Western blot analysis revealed that the protein
synthesis inhibitor inhibited phosphorylation of ERK1/2, p38 and
JNK at the indicated time-points. (E) ALP staining of the
FGF9/inhibitor-treated monolayer BMSCs. After 7 days of osteogenic
culture induction, ALP expression in the FGF9-treated BMSCs was
reduced and this was rescued by the treatment with ERK1/2
inhibitor. However, rescue was not observed in the BMCs treated
with the p38 and JNK inhibitors. The results are representative of
three independent experiments. BMSC, bone marrow stromal stem cell;
p-ERK1/2, phosphorylated extracellular signal-regulated kinase 1/2;
JNK, c-Jun N-terminal kinase; CON, Dulbecco’s modified Eagle’s
medium; OI, osteogenic induction medium; FGF9, fibroblast growth
factor 9; U, 10 μM U0126; SB, 10 μM SB203580; SP, 25 μM SP600125;
ALP, alkaline phosphatase. |
Effects of the FGF9/MAPK signaling
pathway in the osteogenic differentiation of BMSCs in vitro
A previous study by our group demonstrated that FGF9
promotes chondrogenesis and, at the same time, inhibits the
hypotrophy in DPSCs by binding to FGFR3 and enhancing ERK1/2
phosphorylation (13). To
investigate the role of the MAPK pathway in the osteogenic
differentiation of BMSCs, the drugs U0126 (10 μM), SB203580 (10 μM)
and SP600125 (25 μM), which inhibit the phosphorylation of ERK1/2,
p38 and JNK, respectively, were applied and their efficacy was
confirmed by western blot analysis (Fig. 3B–D). Subsequent ALP staining
revealed that exogenous FGF9 inhibited the expression of ALP,
whereas exogenous FGF9 combined with the inhibition of p38 and JNK
phosphorylation had an apparent synergistic effect on the
inhibition of ALP expression in BMSCs (Fig. 3E). By contrast, the inhibition of
ERK1/2 phosphorylation reversed the effect of FGF9 and increased
the expression of ALP (Fig. 3E).
RT-qPCR and western blot analyses further demonstrated that FGF9
treatment decreased the expression of the osteogenic genes COL1 and
Runx2, whereas U0126 reversed the inhibition of the expression of
Runx2, COL1, OPN and OCN (Fig.
4A–D). Western blot analysis of Runx2 and alizarin red staining
further confirmed that inhibition of the osteogenic capacity of
BMSCs by FGF9 was reversed when ERK1/2 phosphorylation was
inhibited (Fig. 4E and F).
 | Figure 4Effect of the FGF9/ERK1 signaling
pathway on the osteogenic differentiation of BMSCs in vitro.
(A–D) mRNA expression of four osteogenic marker genes, Runx2, COL1,
OPN and OCN, in treated BMSCs were investigated using reverse
transcription quantitative polymerase chain reaction. The gene
expression levels of the four osteogenic transcription factors were
downregulated by exogenous FGF9 (20 ng/ml) after 12 h and 7, 14 and
21 days after treatment, respectively. U0126 (10 μM) reversed the
inhibited expression of the four osteogenic marker genes. (E)
Levels of Runx2 protein expression in BMSCs were investigated by
western blot analysis. (F) Alizarin red staining of treated
monolayer BMSCs. Western blot analysis of Runx2 and alizarin red
staining confirmed that the inhibition of the osteogenic capacity
of BMSCs by FGF9 was reversed when ERK1/2 phosphorylation was
inhibited. Values are expressed as the mean ± standard deviation of
triplicate independent experiments.*P<0.05,
**P<0.01. CON, Dulbecco’s modified Eagle’s medium;
OI, osteogenic induction medium; FGF9, 20 ng/ml exogenous
fibroblast growth factor 9; U, 10 μM U0126; Runx2, runt-related
transcription factor 2; BMSCs, bone marrow stromal stem cells;
COL1, collagen type 1; OPN, osteopontin; OCN, osteocalcin. |
Effects of the FGF9/ERK1/2 signaling
pathway in the osteogenic differentiation of DPSCs in vitro
In the present study, the results of western blot
analysis demonstrated that the phosphorylation of ERK1/2 was
augmented in DPSCs cultured in osteogenic medium. The results also
revealed that the incubation of DPSCs cultured in osteogenic medium
including 20 ng/ml FGF9 markedly augmented the phosphorylation of
ERK1/2, beginning at 5 min and gradually increasing thereafter,
whereas 10 μM U0126 effectively inhibited the phosphorylation of
ERK1/2 (Fig. 5A). Similar to the
results observed in the BMSCs, western blot analysis demonstrated
that FGF9 inhibited the expression of COL1 and Runx2 in the DPSCs,
whereas the inhibition of ERK1/2 phosphorylation reversed the
inhibitory effect of FGF9 (Fig.
5B). The results of the ALP and alizarin red staining also
indicated that FGF9 inhibited the expression of ALP and
ossification, which was reversed when combined with the inhibition
of ERK1/2 phosphorylation (Fig.
5C). The RT-qPCR analysis further revealed that FGF9 treatment
decreased the expression of Runx2, COL1, OPN and OCN, whereas U0126
reversed this inhibitory effect and upregulated the expression of
Runx2, COL1, OPN and OCN (Fig.
5D–G).
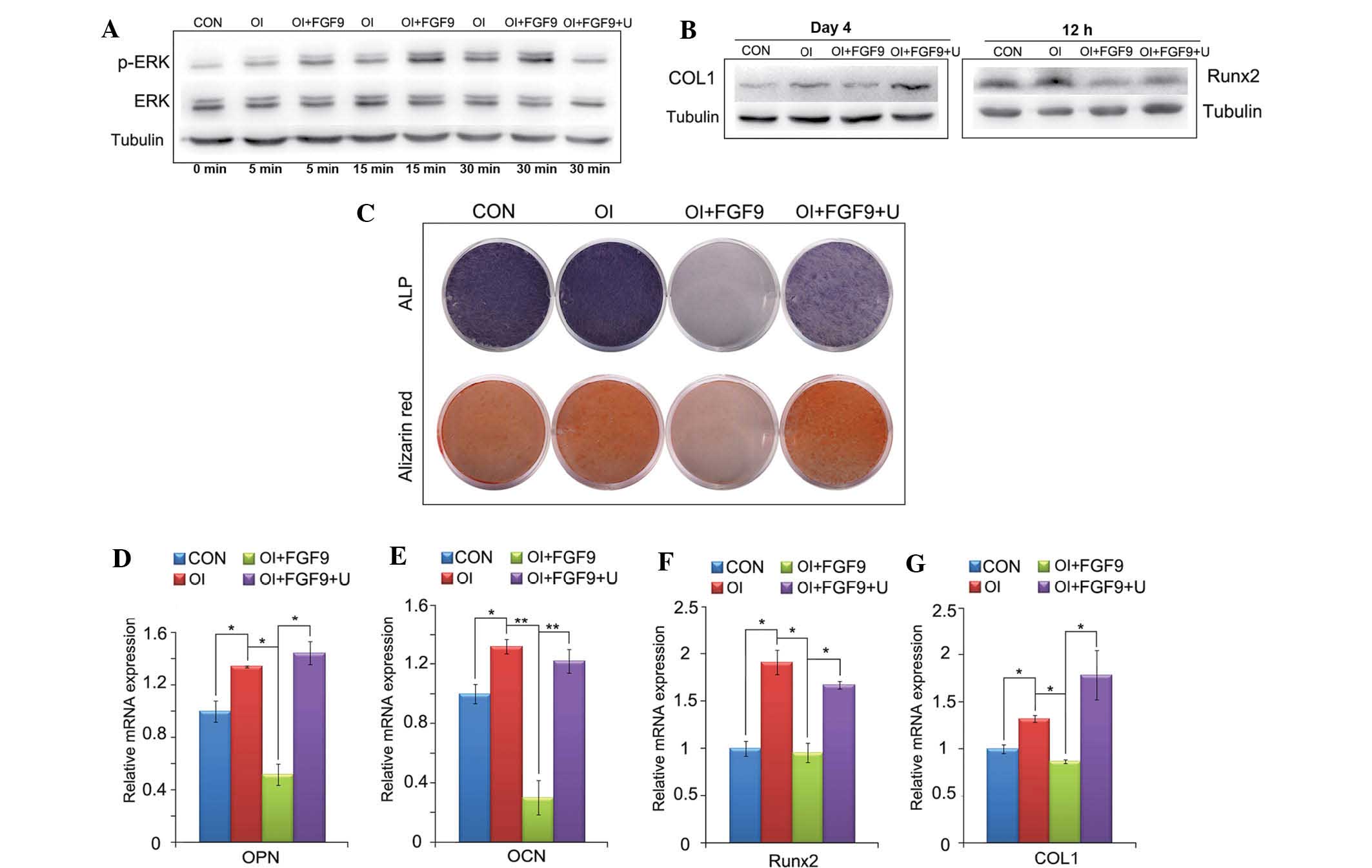 | Figure 5Effect of the FGF9/ERK signaling
pathway on the osteogenic differentiation of DPSCs in vitro.
(A) Expression of ERK1/2 and p-ERK1/2 in treated monolayer DPSCs
was detected by western blot analysis at the indicated time-points.
Tubulin was used to demonstrate equal loading of all samples. FGF9
(20 ng/ml) significantly enhanced the phosphorylation of ERK1/2 in
DPSCs compared with that in the CON and OI groups. (B) Protein
levels of Runx2/COL1 12 h and 4 days after treatment in DPSCs were
investigated by western blot analysis. (C) ALP staining at day 7
and alizarin red staining at day 21 of the FGF9/inhibitor-treated
monolayer DPSCs. Western blot analysis for Runx2 and COL1, ALP and
alizarin red staining revealed that the inhibition of the
osteogenic capacity of DPSCs by FGF9 was reversed when ERK1/2
phosphorylation was inhibited. (D–G) mRNA expression levels of
Runx2, COL1, OPN and OCN in treated DPSCs were investigated using
reverse transcription quantitative polymerase chain reaction. The
gene and protein expression levels of these four osteogenic
transcription factors were downregulated by exogenous FGF9 (20
ng/ml) 12 h and 7, 14 and 21 days after treatment, respectively.
U0126 (10 μM) reversed the inhibited expression of the four
osteogenic marker genes. Data are expressed as the mean ± standard
deviation of triplicate independent experiments.
*P<0.05, **P<0.01. DPSCs, dental pulp
stem cells; CON, Dulbecco’s modified Eagle’s medium; OI, osteogenic
induction medium, FGF9, 20 ng/ml exogenous fibroblast growth factor
9; U, 10 μM U0126; ERK, extracellular signal-regulated kinase;
p-ERK, phosphorylated ERK; Runx2, runt-related transcription factor
2; COL1, collagen type 1; OPN, osteopontin; OCN, osteocalcin; ALP,
alanine phosphatase. |
Discussion
The present study provided evidence that the
FGF9-ERK1/2 signaling pathway is important in the osteogenic
differentiation process in different cell types. The results
revealed that FGF9 inhibited the osteogenic differentiation of
BMSCs and DPSCs in vitro, contradicting previous findings
suggesting that FGF9 is necessary for growth plate development,
osteogenesis and long bone repair in vivo (4,7).
These differences may be attributed to the fact that the BMSCs in
the present in vitro study were directly induced to
differentiate into osteoblasts, whereas previous in vivo
studies focused mainly on endochondral ossification, in which
mesenchymal stem cells first differentiate into chondrocytes and
are then gradually replaced by bone tissue. Previous studies also
demonstrated that FGF9 promotes the differentiation and
chondrogenesis of stem cells. Weksler et al (10) observed that FGF9 led to the
promotion of proliferation of an in vitro rat
calvaria-derived cell line; however, it caused inhibition of its
terminal differentiation. Previous findings by our group revealed
that FGF9 promotes the chondrogenesis and simultaneously inhibits
the hypertrophy of DPSCs (13).
Thus, although FGF9 is able to impair the osteogenic
differentiation of stem cells and inhibit the hypertrophy of
chondrocytes, endochondral ossification remains enhanced due to the
increased cartilage formation. Additionally, a number of other
factors, including bone morphogenetic proteins-2 and 4, promote
osteoblast differentiation and may compensate for the inhibitory
effect of FGF9 on osteogenic differentiation. Several previous
studies have also demonstrated that FGF9 may promote angiogenesis
through the expression of VEGF, thereby promoting bone formation
(4,7,16).
Mice lacking FGF9 have been observed to exhibit reduced chondrocyte
proliferation, delayed chondrocyte hypertrophy and defects in
skeletal vascularization leading to abnormalities in osteogenesis.
These findings suggest that FGF9 regulates skeletal development
in vivo at different stages and that the biological effect
of FGF9 in vivo may depend on gene dosage, spatial and
temporal expression patterns, developmental stage or interaction
with other cytokines. In the present in vitro study, FGF9
had an inhibitory effect on the osteogenic differentiation of BMSCs
and DPSCs. Combined with previous findings by our group that FGF9
simultaneously promotes the chondrogenesis and inhibits the
hypertrophy in DPSCs, this suggested that FGF9 may be applied to
promote the chondrogenesis of mesenchymal stem cells whilst
inhibiting osteogenesis in vitro. As BMSCs originate from
mesoderm, whereas DPSCs may be derived from neural ectoderm
(17), the results of the present
study indicated that the effects of FGF9 on osteogenic
differentiation are similar in stem cells originating from the
mesoderm or ectoderm in vitro. Therefore, potential methods
for the application of FGF9 to promote chondrogenesis in
vivo require investigation in the future.
Previous studies have revealed that the FGF9/FGFR3
signaling pathway activates the ERK1/2 pathway to regulate the
chondrogenic or osteogenic differentiation of BMSCs (12). Several previous studies have also
indicated a complex role for the ERK1/2 signaling pathway during
chondrogenesis and osteogenesis in mesenchymal stem cells (MSCs)
(18). Certain studies have
demonstrated that activation of the ERK/MAPK signaling pathway
promotes chondrogenesis and inhibits hypertrophy in MSCs, whereas
others have demonstrated that inhibition of the ERK/MAPK signaling
pathway promotes osteogenesis in MSCs (19,20).
In the present study, FGF9 enhanced the phosphorylation and
activation of ERK1/2 in BMSCs and DPSCs to inhibit their osteogenic
differentiation. These findings revealed that FGF9 activated
ERK1/2, which was followed by the inhibition of osteogenic
differentiation in BMSCs and DPSCs. These findings were consistent
with those of previous studies, in which decreased phosphorylation
of ERK1/2 promoted the osteogenic differentiation of BMSCs, whereas
enhanced ERK1/2 phosphorylation promoted the osteogenic
differentiation of cranial suture cells, leading to
craniosynostosis (21,22).
In conclusion, the present study revealed that FGF9
enhanced the phosphorylation of ERK1/2 in BMSCs and DPSCs during
osteogenic induction in vitro and inhibited the osteogenic
differentiation of BMSCs and DPSCs through the activation of
ERK1/2. These findings suggested that FGF9 may be an inhibitor of
osteogenesis in MSCs in vitro. However, the in vivo
application of this interaction requires further investigation.
Acknowledgements
This study was supported by the National Nature
Science Foundation of China (nos. 81300842 and 81271122), the
Program for Innovation Research Team of Shanghai Municipal
Education Commission, Shanghai Leading Academic Discipline Project
(no. S30206) and the Clinical and Basic Research on Prevention and
Management of Craniofacial Deformities (Shanghai University
Innovation Team).
References
|
1
|
Sylvestersen KB, Herrera PL, Serup P and
Rescan C: Fgf9 signalling stimulates Spred and Sprouty expression
in embryonic mouse pancreas mesenchyme. Gene Expr Patterns.
11:105–111. 2011. View Article : Google Scholar
|
|
2
|
Geske MJ, Zhang X, Patel KK, Ornitz DM and
Stappenbeck TS: Fgf9 signaling regulates small intestinal
elongation and mesenchymal development. Development. 135:2959–2968.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
White AC, Lavine KJ and Ornitz DM: FGF9
and SHH regulate mesenchymal Vegfa expression and development of
the pulmonary capillary network. Development. 134:3743–3752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hung IH, Yu K, Lavine KJ and Ornitz DM:
FGF9 regulates early hypertrophic chondrocyte differentiation and
skeletal vascularization in the developing stylopod. Dev Biol.
307:300–313. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Govindarajan V and Overbeek PA: FGF9 can
induce endochondral ossification in cranial mesenchyme. BMC Dev
Biol. 6:72006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
DiNapoli L, Batchvarov J and Capel B: FGF9
promotes survival of germ cells in the fetal testis. Development.
133:1519–1527. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Behr B, Leucht P, Longaker MT and Quarto
N: Fgf-9 is required for angiogenesis and osteogenesis in long bone
repair. Proc Natl Acad Sci USA. 107:11853–11858. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ignelzi MA Jr, Wang W and Young AT:
Fibroblast growth factors lead to increased Msx2 expression and
fusion in calvarial sutures. J Bone Miner Res. 18:751–759. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fakhry A, Ratisoontorn C, Vedhachalam C,
et al: Effects of FGF-2/-9 in calvarial bone cell cultures:
differentiation stage-dependent mitogenic effect, inverse
regulation of BMP-2 and noggin, and enhancement of osteogenic
potential. Bone. 36:254–266. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Weksler NB, Lunstrum GP, Reid ES and
Horton WA: Differential effects of fibroblast growth factor (FGF) 9
and FGF2 on proliferation, differentiation and terminal
differentiation of chondrocytic cells in vitro. Biochem J.
342:677–682. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Garofalo S, Kliger-Spatz M, Cooke JL, et
al: Skeletal dysplasia and defective chondrocyte differentiation by
targeted overexpression of fibroblast growth factor 9 in transgenic
mice. J Bone Miner Res. 14:1909–1915. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu XL, Gu MM, Huang L, et al: Multiple
synostoses syndrome is due to a missense mutation in exon 2 of FGF9
gene. Am J Hum Genet. 85:53–63. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dai J, Wang J, Lu J, et al: The effect of
co-culturing costal chondrocytes and dental pulp stem cells
combined with exogenous FGF9 protein on chondrogenesis and
ossification in engineered cartilage. Biomaterials. 33:7699–7711.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ornitz DM and Marie PJ: FGF signaling
pathways in endochondral and intramembranous bone development and
human genetic disease. Genes Dev. 16:1446–1465. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zou D, Zhang Z, He J, et al: Repairing
critical-sized calvarial defects with BMSCs modified by a
constitutively active form of hypoxia-inducible factor-1alpha and a
phosphate cement scaffold. Biomaterials. 32:9707–9718. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Frontini MJ, Nong Z, Gros R, et al:
Fibroblast growth factor 9 delivery during angiogenesis produces
durable, vasoresponsive microvessels wrapped by smooth muscle
cells. Nat Biotechnol. 29:421–427. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Komada Y, Yamane T, Kadota D, et al:
Origins and properties of dental, thymic, and bone marrow
mesenchymal cells and their stem cells. PLoS One. 7:e464362012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Stanton LA, Underhill TM and Beier F: MAP
kinases in chondrocyte differentiation. Dev Biol. 263:165–175.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Prasadam I, van Gennip S, Friis T, Shi W,
Crawford R and Xiao Y: ERK-1/2 and p38 in the regulation of
hypertrophic changes of normal articular cartilage chondrocytes
induced by osteoarthritic subchondral osteoblasts. Arthritis Rheum.
62:1349–1360. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhao Y, Song T, Wang W, et al: P38 and
ERK1/2 MAPKs act in opposition to regulate BMP9-induced osteogenic
differentiation of mesenchymal progenitor cells. PLoS One.
7:e433832012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Twigg SR, Vorgia E, McGowan SJ, et al:
Reduced dosage of ERF causes complex craniosynostosis in humans and
mice and links ERK1/2 signaling to regulation of osteogenesis. Nat
Genet. 45:308–313. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nakamura T, Gulick J, Pratt R and Robbins
J: Noonan syndrome is associated with enhanced pERK activity, the
repression of which can prevent craniofacial malformations. Proc
Natl Acad Sci USA. 106:15436–15441. 2009. View Article : Google Scholar : PubMed/NCBI
|














