Introduction
Millions of people succumb to acute myocardial
infarction (AMI) each year (1),
thus AMI makes a significant contribution to the global burden of
disease. In the past few decades, it was identified that
experiencing short periods of myocardial ischemia and reperfusion
(I/R), prior to restoration of full coronary reperfusion, has a
protective effect on cardiomyocytes against subsequent prolonged
I/R injury; a phenomenon termed ‘ischemic preconditioning’ (IPC)
(2). Further studies have shown
that volatile anesthetics, such as isoflurane, can simulate the
effect of IPC. When administered prior to a period of myocardial
I/R, volatile anesthetics induce cardioprotective effects, which
are referred to as ‘anesthetic preconditioning’ (APC) (3,4). APC
can lead to increased resistance of cardiomyocytes against (I/R)
injury by eliciting endogenous protective mechanisms. This was
observed in various animal models, as well as in humans (3–8). In
contrast to IPC, APC may not cause a reduction in blood flow, thus
it exhibits greater ethical acceptability and clinical safety.
Protein kinase C (PKC)ɛ activation is required to
protect the heart from (I/R) injury (9,10).
Recent evidence suggests that PKCɛ is targeted to the mitochondria
and interacts with numerous mitochondrial proteins, including
mitochondrial aldehyde dehydrogenase 2 (ALDH2) (11). The mitochondrial isoform of ALDH2
is key in the metabolism of acetaldehyde and other toxic aldehydes,
and phosphorylation and activation by PKCɛ are required to confer
cardioprotection (10,11). Overexpression of ALDH2 alleviates
I/R injury, post-I/R injury and ischemic ventricular dysfunction
(12,13). Consistent with this, ALDH2
expression was downregulated during cardiomyocyte hypoxia (14), and ALDH2 knockout exacerbated the
I/R injury (15). These data
support the essential role of ALDH2 in the protection against I/R
injury in the heart. The mechanisms underlying ALDH2-induced
protection against I/R injury are likely to be various and diverse,
involving bioactivation of nitroglycerin; decreasing the production
of free radicals (16) and the
formation of 4-hydroxy-2-nonenal (HNE)-protein adducts (17); the activation of c-Jun N-terminal
kinases 1/2 and extracellular signal-regulated kinases 1/2
(18); and mitochondrial
dysfunction (19–24), which are all hallmarks of I/R
injury. It is suggested that during I/R injury, the overall levels
of PKCɛ and PKCδ are regulated by the proteasome, a multi-subunit
complex found predominantly in the cytosol of mammalian cells,
which can result in the degradation of PKCδ (22,25).
The proteasome regulates the ratio of pro-apoptotic PKCδ to
pro-survival PKCɛ in the mitochondria, and thus determines the
ultimate fate of the cell and may be viewed as an indicator of
cellular viability (22). Uecker
et al (11) reported that
PKCδ was translocated exclusively to the mitochondria in response
to isoflurane treatment, rather than the cell membrane, suggesting
the importance of this isoform in mitochondrial adenosine
triphosphate (ATP)-dependent potassium channel-mediated cardiac
protection by isoflurane. However, a recent report by Xu et
al (39) has shown that APC
increased the levels of PKCɛ and PKCδ in the cell membrane, and
decreased the levels in the cytosol. The role of PKCδ in APC and
the mechanism conferring cardioprotection have not yet been
elucidated. Therefore, the present study aimed to investigate the
role of PKCδ in APC and its underlying mechanism of action.
Materials and methods
Animals
The present study was approved by the Ethics
Committee of Shanxi Medical University (Taiyuan, China). Male
Sprague-Dawley (SD) rats, weighing 200–220 g, were used in this
study. The animals were provided by The Experimental Animal Center
of Tsinghua University (Beijing, China). The rats were placed in a
quiet, temperature- (23±3°C) and humidity- (60±5%) controlled room,
with a 12/12 h light-dark cycle (light beginning at 0800). Rats had
free access to a standard diet and drinking water. Experiments were
performed in accordance with the Guide for the Care and Use of
Laboratory Animals of the China National Institutes of Health.
In vivo I/R injury experimental
protocol
The acute myocardial I/R injury model was performed
by left anterior descending (LAD) coronary artery ligation. Male SD
rats were anesthetized by intraperitoneal administration of 30
mg/kg pentobarbital sodium. After a tracheotomy had been performed,
rat lungs were ventilated mechanically with positive pressure
ventilation using a 30–40% air/oxygen mixture to maintain arterial
blood gas pH within a physiological range by adjusting the
respiratory rate and tidal volume throughout the experiment.
Myocardial infarction (MI) was caused by ligation of the LAD
coronary artery. Briefly, the thorax was opened at the fourth or
fifth left intercostal space. After left thoracotomy and
pericardiotomy, MI was induced by LAD ligation 2–3 mm from the
origin with a 6-0 silk suture (Hairmer Co., Xi’an, China). All
animals (except for the rats in the sham groups) were subjected to
40 min of regional myocardial ischemia followed by 120 min of
reperfusion (26). To confirm
isoflurane-induced APC, a minimal alveolar concentration of
isoflurane of 1.0 (2.1%) was administered at the end of the
stabilization period for 30 min, followed by 30 min of washout with
oxygen prior to coronary occlusion.
Rats were randomly assigned to the following groups
(n=8 per group): Sham group, a non-ischemic control group of
sham-operated rats without isoflurane pretreatment (chest walls
were opened without ligating the LAD coronary artery for 160 min);
non-ischemic control group comprising sham-operated rats pretreated
with isoflurane; I/R group (40 min of myocardial ischemia and 120
min of reperfusion) without isoflurane pretreatment; and I/R group
with isoflurane pretreatment. To evaluate the role of PKCδ in
phosphorylation of ALDH2 and in isoflurane-induced APC, a direct
inhibitor of PKCδ, rottlerin (1 μM), was administered 5 min prior
to ischemia with and without isoflurane to subgroups of the
rats.
Analyzing the activity of lactate
dehydrogenase (LDH) and creatine kinase-MB (CK-MB) in plasma
Serum CK-MB analysis is a widely used biomarker to
detect cardiac injury. Proportionally greater serum CK-MB relative
to the total CK activity can evaluate acute myocardial injury. At
the end of the reperfusion period, 5 ml blood samples were taken.
Serum was separated by centrifugation at 5,000 × g for 5 min on a
tabletop centrifuge and the supernatant was stored in liquid
nitrogen. The samples were thawed for analysis. LDH and CK-MB were
assayed using LDH and CK-MB commercially available kits (Roche,
Manheim, Germany), respectively, by an automatic analyzer 7600
(Hitachi, Tokyo, Japan).
Preparation of whole cell extracts from
myocardium and western blot analysis
To identify the effect of isoflurane preconditioning
on PKCδ activation and translocation, mito-PKCδ and total-PKCδ
expression in all groups was measured (Sham, I/R, Sham+isoflurane
and I/R+isoflurane groups) by western blot analysis. The effect of
rottlerin on signal pathway proteins (phos-ALDH2 and total-ALDH2)
was also assayed by western blot analysis.
Upon completion of the experimental period, the
myocardium and cardiomyocytes were lysed in ice-cold
radioimmunoprecipitation assay lysis buffer containing 1 mmol/l
phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin, 1 μg/ml aprotinin
and 1 μg/ml pepstatin at 4°C for 15 min. The homogenate was
incubated and centrifuged at 5,000 × g for 5 min at 4°C. The
supernatant was collected and the protein concentration was
determined using the bicinchoninic acid protein assay kit (Pierce
Biotechnology Inc., Rockford, IL, USA) according to the
manufacturer’s instructions. The detergent soluble supernatant was
frozen with liquid N2 and stored at −70°C.
The supernatant was mixed with 5X loading buffer and
heated for 5 min at 100°C. Soluble extracts (50 μg) were loaded in
each lane and separated by SDS-polyacrylamide gel electrophoresis.
Following electrophoresis, proteins were electrophoretically
transferred to a polyvinylidene difluoride filter membrane (0.45
μm, GE Healthcare, Beijing, China). The membrane was blocked in
Tris-buffered saline with Tween-20 (TBST) with 5% non-fat milk and
incubated overnight with the corresponding primary antibodies at
4°C. The following primary antibodies were used: Rabbit monoclonal
anti-PKCδ and rabbit monoclonal PKCɛ (both Abcam, Cambridge, UK).
The membrane was then incubated for 1 h with secondary antibody
horseradish peroxidase-conjugated goat anti-rabbit IgG (Beyotime
Institute of Biotechnology, Haimen, China) diluted with TBST
(1:2,000). The signals of detected proteins were visualized by an
enhanced chemiluminescence reaction system (Millipore, Billerica,
MA, USA). The staining was quantified by scanning the films and the
band density was determined with Image-Pro software (Media
Cybernetics, Inc., Rockville, MA, USA).
Statistical analysis
Continuous values are expressed as the mean ±
standard error of the mean. Comparisons between multiple-group
means were performed using one-way analysis of variance and
comparisons between groups were performed using the least
significant difference test and Student-Newman-Keuls test. The
number of animals per group and statistical significance for all
data are listed in the figures and figure legends. P<0.05 was
considered to indicate a statistically significant difference. All
statistical analyses were performed using SPSS version 15.0 (SPSS
Inc., Chicago, IL, USA).
Results
Effect of isoflurane pretreatment on PKCδ
activity and mitochondrial PKCδ levels
Regional myocardial ischemia for 40 min by LAD
coronary artery ligation followed by 120 min of reperfusion led to
a significant increase in PKCδ levels in the mitochondria of
cardiomyocytes compared with the sham control group (P<0.01).
Isoflurane markedly inhibited the I/R-induced mitochondrial
translocation of PKCδ in cardiomyocytes, and significantly
decreased the mitochondrial PKCδ concentration in cardiomyocytes
(P<0.01; Fig. 1A and B). Data
suggested that pretreatment with isoflurane decreased the dynamic
mitochondrial translocation of PKCδ in response to I/R.
As shown in Fig.
1A, the decrease in the mitochondrial concentration of PKCδ in
cardiomyocytes indicated the translocation of PKCδ from the
mitochondria to the cytosol, and the corresponding increase in
cytosolic PKCδ levels during isoflurane pretreatment, as the total
cellular PKCδ levels remained constant.
Role of PKCδ in isoflurane-induced ALDH2
phosphorylation and cardioprotection
PKCδ is involved in isoflurane-induced ALDH2
phosphorylation and cardioprotection. PKCδ activation at the
beginning of reperfusion mediates cardiomyocyte apoptosis and
necrosis via mitochondrial regulation, whilst PKCδ inhibition
alleviates the myocardial I/R injury (27). Decreased levels of PKCδ and
elevated levels of phosphorylation of ALDH2 are required to protect
the heart from I/R injury. Isoflurane-induced ALDH2 phosphorylation
level decrease (P<0.01, Fig. 2A and
B). LDH (P<0.05) and CK-MB (P<0.01) release (Fig. 2C and D) induced by I/R was mimicked
by the PKCδ inhibitor, rottlerin. Western blot analysis showed that
inhibition of PKCδ activity by rottlerin after I/R injury
significantly enhanced the phosphorylation level of ALDH2
regardless of isoflurane preconditioning. Consequently, the
inhibition of PKCδ was associated with ALDH2 activation and was
observed to induce cardioprotection, demonstrated by decreased
serum CK-MB and LDH activity in vivo (Fig. 2B–D). Similarly, isoflurane
preconditioning inhibited PKCδ activity, thus inhibiting its
mitochondrial translocation and reducing I/R-induced myocardial
injury. A significant difference was observed between the group
treated with rottlerin alone and the group that received
co-treatment with rottlerin and isoflurane in terms of LDH and
CK-MB release (P<0.01), which indicates a synergistic effect on
decreasing the two biochemical indicators (Fig. 2C and D). However, no such effect on
ALDH2 phosphorylation was observed (Fig. 2A and B).
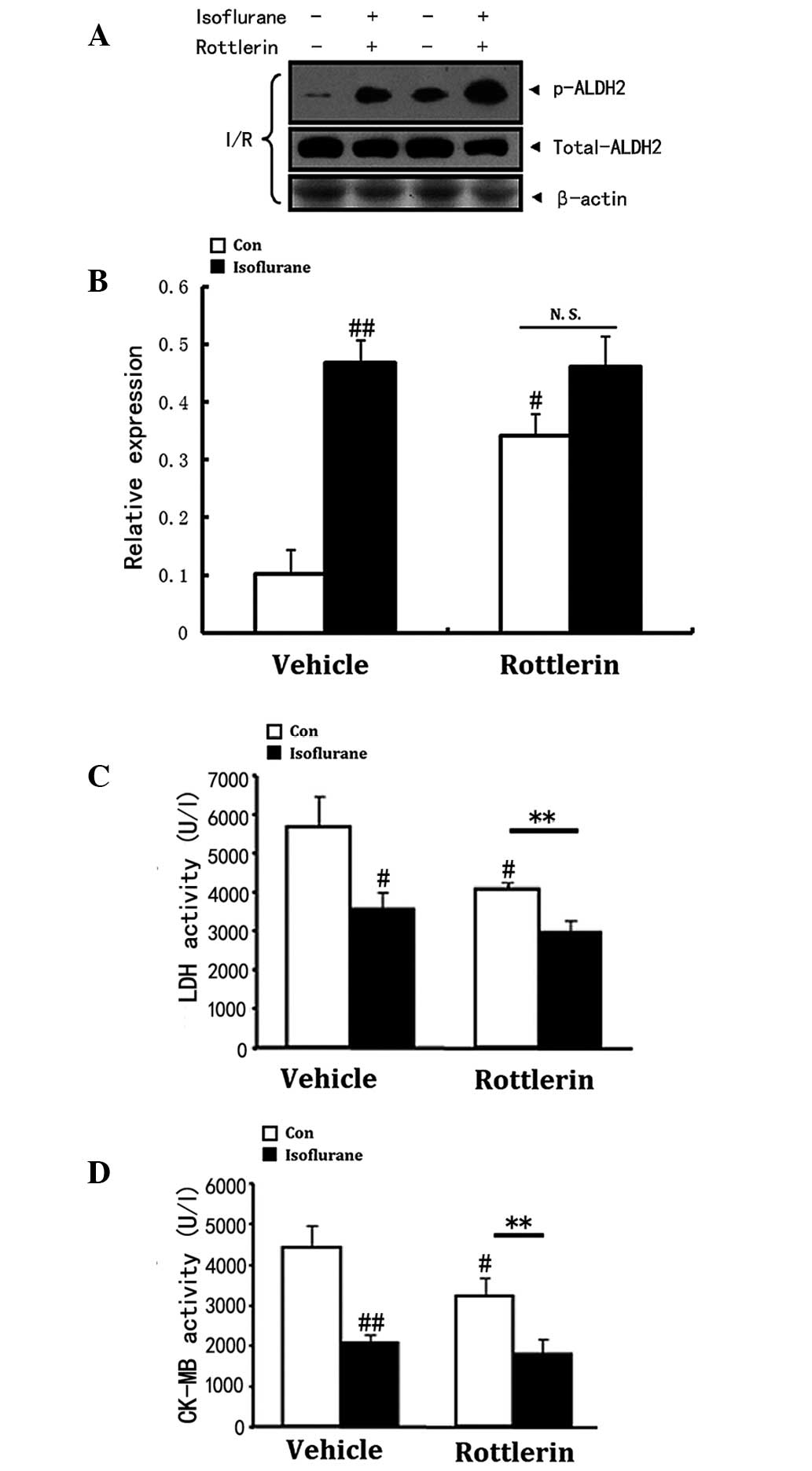 | Figure 2Decrease in PKCδ translocation to the
mitochondria involves ALDH2 phosphorylation and cardioprotection
induced by isoflurane. Values are presented as the mean ± standard
error of the mean, n=8 per group. #P<0.05,
##P<0.01 vs. the vehicle control group and
**P<0.01 vs. the corresponding control. (A and B) The
roles of PKCδ in isoflurane preconditioning induced ALDH2
activation. Representative western blot of phospho-ALDH2, total
ALDH2 after I/R injury. Inhibition of PKCδ significantly increased
the phosphorylation of ALDH2 regardless of isoflurane
preconditioning. β-actin was used to demonstrate equal protein
loading. (C and D) Serum LDH and CK-MB concentrations were
analyzed. Isoflurane inhibited LDH and CK-MB release induced by
I/R, which was mimicked by rottlerin (a PKCδ inhibitor). Serum LDH
and CK-MB concentrations were analyzed. There was no significant
difference between the single rottlerin treatment group, and the
rottlerin and isoflurane co-treatment group in ALDH2 relative
expression, which suggested the effects of isoflurane and rottlerin
were not synergistic. In terms of the LDH and CK-MB levels, there
was a significant difference between the single rottlerin treatment
group, and the rottlerin and isoflurane co-treatment group,
indicating a synergistic effect of isoflurane and rottlerin. ALDH2,
aldehyde dehydrogenase 2; PKC, protein kinase C; I/R,
ischemia/reperfusion; LDH, lactate dehydrogenase; CK-MB, creatine
kinase-MB. |
Discussion
Clinically, volatile anesthetics have been in use
for a considerable period of time. A number of studies, in
agreement with the current study, have demonstrated the
cardioprotective effects of volatile anesthetics applied before a
deleterious ischemic event and at the beginning of reperfusion,
which share common characteristics with IPC.
However, the rapid induction of pro-survival
pathways sufficient to prevent damage immediately after the
reperfusion event occurs, is likely to be difficult to achieve in
practice. Recently, attention has paid to mitochondria as a target
of volatile anesthetics when inducing cardioprotection (11,28,29).
Mitochondria are fully involved in the pathways induced by volatile
anesthetics, leading to the cardioprotective effects via production
of ATP and regulation of cell death (30).
The mechanisms by which isoflurane ultimately limits
infarct size are not known. Apoptosis (31–33)
and inflammation (31,34) have been implicated in cardiac I/R
injury. In agreement with our previous results (35), isoflurane-treated mice subjected to
ischemia and 2 weeks of reperfusion showed lower expression of
proapoptotic genes, significantly decreased expression of cleaved
caspase-3 and significantly decreased TUNEL staining, as compared
with the control group (5).
ALDH2 is best known for its role in metabolizing the
ethanol intermediate acetaldehyde, which is a toxic aldehyde with
such high activity that it can react with protein, forming
aldehydic adducts and leading to protein dysfunction and tissue
injury. It has been reported that overexpression of ALDH2 may
alleviate I/R injury, post-I/R injury and ischemic ventricular
dysfunction (36). Consistent with
this, I/R injury may be exacerbated by ALDH2 knockout (37). These data support the results of
the present study, which demonstrate that ALDH2 is essential in
isoflurane-induced cardioprotection against I/R injury. It has been
shown that overexpression of ALDH2 significantly attenuated
acetaldehyde- and ethanol-induced oxidative stress (ROS
generation), activation of stress signal molecules and apoptosis in
fetal human cardiomyocytes (36).
There are various perspectives regarding the role of
PKCδ in cardioprotection. The present results showed that during
I/R injury, PKCδ translocates from the cytosol to the mitochondria,
which suggests that PKCδ may mediate I/R injury by interacting with
the mitochondria, consistent with previous studies (38). However, no significant difference
was identified in the total-PKCδ levels between the control and
isoflurane groups. This seems to contradict the proteasome
regulation mechanism suggested by Churchill et al (22). Furthermore, it was identified that
following the inhibition of PKCδ, the level of ALDH2
phosphorylation is upregulated, which indicates that PKCδ is
involved in the cardioprotective effect induced by isoflurane via
the ALDH2 pathway. Thus in the PKC signaling pathway, the roles of
PKCɛ and PKCδ are opposing. The former enhances the phosphorylation
and activation of ALDH2, causing cardiac protection, while the
latter attenuates the effect. This conclusion is in accordance with
several previous studies (39).
In our previous studies it was observed that PKCɛ
activation was involved in isoflurane pretreatment, which
consequently activated downstream signaling pathways and aided
cardioprotection (35). Our
previous studies also identified an anti-apoptotic effect mediated
by PKCɛ in isoflurane preconditioning, demonstrated by decreased
caspase-3 activity and apoptotic cell number in vitro. This
suggested that PKCɛ may exhibit a key role in apoptosis inhibition
during cardioprotection (35). By
contrast, the association between PKCδ and apoptosis has also been
reported. PKCδ was shown to be activated by various apoptotic
stimulants and further translocated to the mitochondria, the Golgi
apparatus and the nucleus, causing multiple biological effects
(40–42). Emoto et al (43,44)
observed the cleavage of PKCδ during apoptosis into catalytic
products by caspase-3 (45).
Leverrier et al (46)
reported that overexpression of PKCδ catalytic segments induced
PARP cleavage, which activated caspase-3. It was further suggested
that a positive feedback cycle between PKCδ and caspase-3 was
involved in apoptosis, although the mechanisms remained unclear
(46,47). The findings regarding the roles of
PKCδ phosphorylation and translocation in I/R injury are in
accordance with previous studies, thus it is plausible to suggest
that the two subtypes of PKC (PKCɛ and PKCδ) have different roles
in myocardial I/R following isoflurane pretreatment, effecting
ALDH2 phosphorylation and mitochondrial translocation. This
eventually leads to inhibition of the apoptotic signaling pathway,
in which caspase-3 is involved, and thereby contributes to
cardioprotection.
PKCɛ has been demonstrated to be a critical protein
kinase in ALDH2 activation, whereas few studies have elucidated the
role of PKCδ in regulating ALDH2. To the best of our knowledge, our
previous study (35) showed for
the first time that ALDH2 phosphorylation is the crucial step in
the anti-apoptotic effect mediated by isoflurane pretreatment
protecting against I/R injury in rat cardiomyocytes in vivo.
ALDH2 was the key factor in the protective effect and ALDH2
inhibition eliminated the effect.
In conclusion, to the best of our knowledge the
present study showed for the first time that isoflurane
pretreatment resulted in significantly elevated mitochondrial
levels of PKCɛ accompanied by phosphorylation of ALDH2, as well as
attenuated mitochondrial translocation of PKCδ. The bi-directional
regulation of the activation of the two PKC subtypes during I/R may
further activate ALDH2 and strengthen resistance to I/R injury
(Fig. 3). Thus, isoflurane
preconditioning may activate the PKC (PKCɛ and PKCδ)-ALDH2
signaling pathway to induce protective effects against myocardial
I/R injury. This study may facilitate the application of APC to
induce cardioprotection in the clinical setting.
References
|
1
|
Keeley EC, Boura JA and Grines CL: Primary
angioplasty versus intravenous thrombolytic therapy for acute
myocardial infarction: a quantitative review of 23 randomised
trials. Lancet. 361:13–20. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Murry CE, Jennings RB and Reimer KA:
Preconditioning with ischemia: a delay of lethal cell injury in
ischemic myocardium. Circulation. 74:1124–1136. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kersten JR, Schmeling TJ, Pagel PS, Gross
GJ and Warltier DC: Isoflurane mimics ischemic preconditioning via
activation of K(ATP) channels: reduction of myocardial infarct size
with an acute memory phase. Anesthesiology. 87:361–370. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cason BA, Gamperl AK, Slocum RE and Hickey
RF: Anesthetic-induced preconditioning: previous administration of
isoflurane decreases myocardial infarct size in rabbits.
Anesthesiology. 87:1182–1190. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tsutsumi YM, Patel HH, Lai NC, Takahashi
T, Head BP and Roth DM: Isoflurane produces sustained cardiac
protection after ischemia-reperfusion injury in mice.
Anesthesiology. 104:495–502. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Toller WG, Kersten JR, Pagel PS, Hettrick
DA and Warltier DC: Sevoflurane reduces myocardial infarct size and
decreases the time threshold for ischemic preconditioning in dogs.
Anesthesiology. 91:1437–1446. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Penta de Peppo A, Polisca P, Tomai F, et
al: Recovery of LV contractility in man is enhanced by preischemic
administration of enflurane. Ann Thorac Surg. 68:112–118. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
De Hert SG, ten Broecke PW, Mertens E, et
al: Sevoflurane but not propofol preserves myocardial function in
coronary surgery patients. Anesthesiology. 97:42–49. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pravdic D, Mio Y, Sedlic F, et al:
Isoflurane protects cardiomyocytes and mitochondria by immediate
and cytosol-independent action at reperfusion. Br J Pharmacol.
160:220–232. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Novalija E, Kevin LG, Camara AK, Bosnjak
ZJ, Kampine JP and Stowe DF: Reactive oxygen species precede the
epsilon isoform of protein kinase C in the anesthetic
preconditioning signaling cascade. Anesthesiology. 99:421–428.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Uecker M, Da Silva R, Grampp T, Pasch T,
Schaub MC and Zaugg M: Translocation of protein kinase C isoforms
to subcellular targets in ischemic and anesthetic preconditioning.
Anesthesiology. 99:138–147. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tanaka K, Weihrauch D, Kehl F, et al:
Mechanism of preconditioning by isoflurane in rabbits: a direct
role for reactive oxygen species. Anesthesiology. 97:1485–1490.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Müllenheim J, Ebel D, Frässdorf J, Preckel
B, Thämer V and Schlack W: Isoflurane preconditions myocardium
against infarction via release of free radicals. Anesthesiology.
96:934–940. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pain T, Yang XM, Critz SD, et al: Opening
of mitochondrial K(ATP) channels triggers the preconditioned state
by generating free radicals. Circ Res. 87:460–466. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kersten JR, Schmeling TJ, Hettrick DA,
Pagel PS, Gross GJ and Warltier DC: Mechanism of myocardial
protection by isoflurane. Role of adenosine triphosphate-regulated
potassium (KATP) channels. Anesthesiology. 85:794–807; discussion
727A. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dana A, Skarli M, Papakrivopoulou J and
Yellon DM: Adenosine A(1) receptor induced delayed preconditioning
in rabbits: induction of p38 mitogen-activated protein kinase
activation and Hsp27 phosphorylation via a tyrosine kinase- and
protein kinase C-dependent mechanism. Circ Res. 86:989–997. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mitchell MB, Meng X, Ao L, Brown JM,
Harken AH and Banerjee A: Preconditioning of isolated rat heart is
mediated by protein kinase C. Circ Res. 76:73–81. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fryer RM, Wang Y, Hsu AK and Gross GJ:
Essential activation of PKC-delta in opioid-initiated
cardioprotection. Am J Physiol Heart Circ Physiol. 280:H1346–H1353.
2001.PubMed/NCBI
|
|
19
|
Ping P, Takano H, Zhang J, et al:
Isoform-selective activation of protein kinase C by nitric oxide in
the heart of conscious rabbits: a signaling mechanism for both
nitric oxide-induced and ischemia-induced preconditioning. Circ
Res. 84:587–604. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cope DK, Impastato WK, Cohen MV and Downey
JM: Volatile anesthetics protect the ischemic rabbit myocardium
from infarction. Anesthesiology. 86:699–709. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Toller WG, Montgomery MW, Pagel PS,
Hettrick DA, Warltier DC and Kersten JR: Isoflurane-enhanced
recovery of canine stunned myocardium: role for protein kinase C?
Anesthesiology. 91:713–722. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Churchill EN and Mochly-Rosen D: The roles
of PKCdelta and epsilon isoenzymes in the regulation of myocardial
ischaemia/reperfusion injury. Biochem Soc Trans. 35:1040–1042.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhong L and Su JY: Isoflurane activates
PKC and Ca2+-calmodulin-dependent protein kinase II via
MAP kinase signaling in cultured vascular smooth muscle cells.
Anesthesiology. 96:148–154. 2002. View Article : Google Scholar
|
|
24
|
Ono Y, Fujii T, Ogita K, Kikkawa U,
Igarashi K and Nishizuka Y: The structure, expression, and
properties of additional members of the protein kinase C family. J
Biol Chem. 263:6927–6932. 1988.PubMed/NCBI
|
|
25
|
Kukan M: Emerging roles of proteasomes in
ischemia-reperfusion injury of organs. J Physiol Pharmacol.
55:3–15. 2004.PubMed/NCBI
|
|
26
|
Raphael J, Rivo J and Gozal Y:
Isoflurane-induced myocardial preconditioning is dependent on
phosphatidylinositol-3-kinase/Akt signalling. Br J Anaesth.
95:756–763. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen CH, Budas GR, Churchill EN, Disatnik
MH, Hurley TD and Mochly-Rosen D: Activation of aldehyde
dehydrogenase-2 reduces ischemic damage to the heart. Science.
321:1493–1495. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mio Y, Bienengraeber MW, Marinovic J, et
al: Age-related attenuation of isoflurane preconditioning in human
atrial cardiomyocytes: roles for mitochondrial respiration and
sarcolemmal adenosine triphosphate-sensitive potassium channel
activity. Anesthesiology. 108:612–620. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ljubkovic M, Mio Y, Marinovic J, et al:
Isoflurane preconditioning uncouples mitochondria and protects
against hypoxia-reoxygenation. Am J Physiol Cell Physiol.
292:C1583–C1590. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hu ZY and Liu J: Mechanism of cardiac
preconditioning with volatile anaesthetics. Anaesth Intensive Care.
37:532–538. 2009.PubMed/NCBI
|
|
31
|
Suzuki K, Murtuza B, Smolenski RT, et al:
Overexpression of interleukin-1 receptor antagonist provides
cardioprotection against ischemia-reperfusion injury associated
with reduction in apoptosis. Circulation. 104:I308–I313. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fliss H and Gattinger D: Apoptosis in
ischemic and reperfused rat myocardium. Circ Res. 79:949–956. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gottlieb RA, Burleson KO, Kloner RA,
Babior BM and Engler RL: Reperfusion injury induces apoptosis in
rabbit cardiomyocytes. J Clin Invest. 94:1621–1628. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Meldrum DR, Dinarello CA, Shames BD, et
al: Ischemic preconditioning decreases postischemic myocardial
tumor necrosis factor-alpha production. Potential ultimate effector
mechanism of preconditioning. Circulation. 98:II214–II219.
1998.PubMed/NCBI
|
|
35
|
Lang XE, Wang X, Zhang KR, Lv JY, Jin JH
and Li QS: Isoflurane preconditioning confers cardioprotection by
activation of ALDH2. PLoS One. 8:e524692013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li SY, Li Q, Shen JJ, et al: Attenuation
of acetaldehyde-induced cell injury by overexpression of aldehyde
dehydrogenase-2 (ALDH2) transgene in human cardiac myocytes: role
of MAP kinase signaling. J Mol Cell Cardiol. 40:283–294. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Endo J, Sano M, Katayama T, et al:
Metabolic remodeling induced by mitochondrial aldehyde stress
stimulates tolerance to oxidative stress in the heart. Circ Res.
105:1118–1127. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zheng H, Liu J, Liu C, et al:
Calcium-sensing receptor activating phosphorylation of PKCδ
translocation on mitochondria to induce cardiomyocyte apoptosis
during ischemia/reperfusion. Mol Cell Biochem. 358:335–343. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hunter JC, Kostyak JC, Novotny JL, Simpson
AM and Korzick DH: Estrogen deficiency decreases ischemic tolerance
in the aged rat heart: Roles of PKCdelta, PKCepsilon, Akt, and
GSK3beta. Am J Physiol Regul Integr Comp Physiol. 292:R800–R809.
2007. View Article : Google Scholar
|
|
40
|
Majumder PK, Mishra NC, Sun X, et al:
Targeting of protein kinase C delta to mitochondria in the
oxidative stress response. Cell Growth Differ. 12:465–470.
2001.PubMed/NCBI
|
|
41
|
Kajimoto T, Ohmori S, Shirai Y, Sakai N
and Saito N: Subtype-specific translocation of the delta subtype of
protein kinase C and its activation by tyrosine phosphorylation
induced by ceramide in HeLa cells. Mol Cell Biol. 21:1769–1783.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Blass M, Kronfeld I, Kazimirsky G,
Blumberg PM and Brodie C: Tyrosine phosphorylation of protein
kinase Cdelta is essential for its apoptotic effect in response to
etoposide. Mol Cell Biol. 22:182–195. 2002. View Article : Google Scholar
|
|
43
|
Emoto Y, Kisaki H, Manome Y, Kharbanda S
and Kufe D: Activation of protein kinase Cdelta in human myeloid
leukemia cells treated with 1-beta-D-arabinofuranosylcytosine.
Blood. 87:1990–1996. 1996.PubMed/NCBI
|
|
44
|
Emoto Y, Manome Y, Meinhardt G, et al:
Proteolytic activation of protein kinase C delta by an ICE-like
protease in apoptotic cells. EMBO J. 14:6148–6156. 1995.PubMed/NCBI
|
|
45
|
Ghayur T, Hugunin M, Talanian RV, et al:
Proteolytic activation of protein kinase C delta by an ICE/CED
3-like protease induces characteristics of apoptosis. J Exp Med.
184:2399–2404. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Leverrier S, Vallentin A and Joubert D:
Positive feedback of protein kinase C proteolytic activation during
apoptosis. Biochem J. 368:905–913. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Basu A and Akkaraju GR: Regulation of
caspase activation and cis-diamminedichloroplatinum(II)-induced
cell death by protein kinase C. Biochemistry. 38:4245–4251. 1999.
View Article : Google Scholar : PubMed/NCBI
|
















