Introduction
Pituitary tumors are derived from highly
differentiated pituitary cell subtypes which are somatotrophs
secreting hormones, lactotrophs expressing prolactin (PRL),
thyrothrophs secreting thyrotropin and corticotrophs secreting
adrenocorticotropin (1).
PRL-producing adenomas are the most common of these.
Pituitary-type adenomas may cause significant local
compression due to the mass effect and inappropriate secretion of
pituitary hormones (2,3). They are commonly encountered and are
invariably benign. Stable or indolent growth has been observed in
pituitary adenomas (4). Pituitary
adenomas rarely progress to malignant carcinoma (5,6). Low
mitotic activity has been observed in aggressive pituitary adenomas
in comparison with tumors arising from more rapidly replicating
tissues (7). A new finding is that
pituitary cells undergo premature senescence, enabling escape from
the proliferative pressure of oncogenes, hormones and
transformation factors (8).
Cellular senescence is broadly considered as the causative
mechanism underlying the benign biological behavior of pituitary
tumors. Cellular senescence is a key biological progress which
controls cell proliferation and may play an essential role in tumor
suppression (8). Age-related
telomere shortening may cause cell senescence, also known cellular
aging or replicate aging (9–11).
By contrast, premature senescence occurring prior to telomere
shortening is caused by oxidative stress, DNA damage, aneuploidy,
chromosomal instability and oncogene activation (12). It is reported that both
non-tumorous and tumorous pituitary cells are prone to senescence
(13,14). Premature cellular senescence is
considered to be the essential mechanism underlying the ability of
pituitary cells to escape aggressive proliferation and malignant
transformation (14,15).
Pituitary cellular senescence is characterized by
increased senescence-associated β-galactosidase (SA-β-Gal)
activity, chromosome condensation, DNA damage and upregulation of
cell cycle progression inhibitors including p19, p16 and p21
(16). The mechanisms underlying
pituitary senescence are not entirely clear. Multiple cellular
pathways and cytokines may be involved in triggering pituitary cell
senescence. One mechanism may be the activation of DNA damage and
p53/p21 senescence induced by the securin properties of the
pituitary tumor transforming gene (PTTG) (17). The pathway of p16/Rb or IL-6/C/EBPβ
mediates the oncogene-induced senescence (18,19).
Autocrine IL-6 promotes cell growth but its paracrine activity is
required for the execution of oncogene-induced senescence (20). The PTTG protein, initially isolated
from pituitary tumor cells, is a mammalian securin which
facilitates sister-chromatid separation during metaphase. PTTG
exhibits oncogene properties as its overexpression promotes cell
malignant transformation and tumor formation (21). PTTG deletion results in pituitary
cell senescence. However, high levels of PTTG may trigger
oncogene-induced senescence (14).
In the present study, we detected the expression of
the proteins involved in premature senescence pathways in normal,
aging and tumorous pituitary gland tissues of rats. We successfully
induced the aging pituitary gland using D-galactose (D-gal)
injection as well as pituitary adenoma by chronic estrogen stress.
Significant upregulation of IL-6, C/EBPβ, p53, p21 and p16
expression as well as decreased expression of PTTG was observed in
aging pituitary tissues. The expression of IL-6, p21 and p16 was
decreased in the pituitary tumor gland compared with normal
pituitary tissues, although there was no difference in the
expression of p53 and C/EBPβ between the tumor and normal groups.
Our data further confirm the senescence-associated gene expression
reported by others and reveal the relative expression of these
genes in normal, aging and neoplasia pituitary glands.
Materials and methods
Animal model
Fischer 344 rats were randomly divided into three
groups with each group consisting of 12 rats. Group A formed the
control group, group B formed the aging group and group C formed
the tumor group. The rats in group C received chronic estrogen
treatment using subcutaneous silastic controlled-release tube
implants filled with 10 mg diethylstilbestrol (DES) as described by
Lloyd (22). Silastic capsules
filled with normal saline were implanted as controls in groups A
and B. The rats in group B were treated with a daily
intraperitoneal injection of D-gal solution at a dose of 200 mg/kg.
The rats were sacrificed by decapitation after 8 weeks. Blood was
collected for PRL and IL-6 assays. The weights of the pituitary and
body were recorded and analyzed. The standards of housing and care
for laboratory animals conformed to the standards in the NIH
Guidelines for the Care and Use of Animals. The experiments were
approved by the Ethics Committee of Soochow University, China.
Immunohistochemistry
Pituitary tissues from the 12 animals in each group
were fixed in Bouin’s fluid and embedded in paraffin. PRL
expression in pituitary tissue was determined using mouse
monoclonal PRL antibody (murine; Dako, Carpinteria, CA, USA). The
sections were immunostained and then incubated for 2 h at room
temperature with anti-PRL primary antiserum, diluted 1:300. The
sections were then treated for 30 min with a ready-to-use EnVision
reaction system (Dako). The peroxide-sensitive chromogen was
diaminobenzidine. Digital images were acquired from the light
microscope (Olympus BX50, Japan) at ×200 magnification via a CCD
color camera (DS-Fi1; Nikon, Japan) and analyzed using a computer
system (NIS-Elements F 3.0; Nikon).
SA-β-Gal staining
SA-β-Gal enzymatic activity was detected using a
staining kit (Genmed Scientifics, Shanghai, China). Briefly,
pituitary tissue samples were snap-frozen at optimal cutting
temperature. Specimens were sliced in 10-μm sections and dried for
1 h at room temperature. The sections were fixed and stained
according to the instruction of manufacturer. The sections were
then visualized by microscopy (Leica, Solms, Germany).
Enzyme-linked immunosorbent assay
(ELISA)
ELISA kits for rat PRL (code no. CSB-E06881r) and
IL-6 (code no. CSB-E04640r) were purchased from Cusabio Company
(Cusabio, Wuhan, China). Plasma was anticoagulated by EDTA and then
centrifuged for 15 min at 1,000 × g. The assay was performed
according to the manufacturer’s instructions.
RNA extraction and quantitative
polymerase chain reaction (qPCR)
Total RNA was extracted using TRIzol reagent
(Invitrogen, Paisley, UK) according to the manufacturer’s
instructions. cDNA was synthesized using random primers (Takara,
Dalian, China) with Moloney murine leukemia virus reverse
transcriptase (Promega, Madison, WI, USA) at 37°C for 1 h. For
qPCR, the specific oligonucleotide primers were as follows: p53,
5′-GCTGAGTATCTGGACGACAGG-3′ and 5′-AGCGTGATGATGGTAAGGATG-3′; p21,
5′-AGGGCTTTCTTTGTGTATTTGC-3′ and 5′-GCCTGTTTCGTGTCTACTGTTC-3′; p16,
5′-GGGCTTCCTAGACACTCTGGTA-3′ and 5′-AGAAGTTATGCCTGTCGGTGAC-3′;
C/EBPβ, 5′-TTGTTGCTGTTGATGTTTTTGTT-3′ and
5′-TCTTCACTTTAATGCTCGAAACG-3′; PTTG, 5′-AAGGCTCTGGGAACTGTCAAC-3′
and 5′-ATTTCTGGGTAGGCATCATCAG-3′; IL-6, 5′-CACAAGTCCGGAGAGGAGAC-3′
and 5′-ACAGTGCATCATCGCTGTTC-3′; and Rps16 which was used as the
internal control, 5′-AAGTCTTCGGACGCAAGAAA-3′ and
5′-TGCCCAGAAGCAGAACAG-3′. qPCR was performed in a 20 μl reaction
volume containing 2 μl cDNA, 2 μl primers, 6 μl double-distilled
H2O and 10 μl SYBR-Green mix (Roche Molecular
Biochemicals, Indianapolis, IN, USA). The PCR protocol consisted of
1 min denaturation at 95°C, 30 sec annealing of primers at 60°C and
30 sec elongation at 72°C, for a total of 30 cycles. Quantification
of PCR was calculated as the difference between the cycle threshold
(Ct) value of the gene and the Ct value of the reference gene
Rps16.
Statistic analysis
The values were presented as the means ± SD. The
results for the groups were compared using Student’s t-test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Effects of DES and D-gal treatment on the
body and pituitary weight of rats
DES implantation and D-gal injection resulted in a
slight decrease in the body weight of the rats in the aging and
tumor groups compared with those in the control group (Fig. 1A). Correspondingly, slow response
and dull fur were observed in the rats treated with D-gal, while
the tumor group demonstrated a notable increase in the discharge of
urine.
Compared with the control group, the pituitary
weight was significantly increased in the tumor group which
received the DES treatment (P<0.01; Fig. 1B–D). In contrast, the pituitary
weights of the aging group treated by D-gal injection were almost
the same as those of the control group (Fig. 1B–D).
Induction of prolactin-secreting
pituitary tumor and aging rats
To further investigate whether DES implantation
induced the pituitary tumor and D-gal treatment promoted pituitary
aging, histological staining of the pituitary gland tissue was
performed. Hematoxylin and eosin staining revealed hypertrophied
cells with large nuclei and structural disorder in the tumor group
receiving DES administration. Correspondingly, strong positive PRL
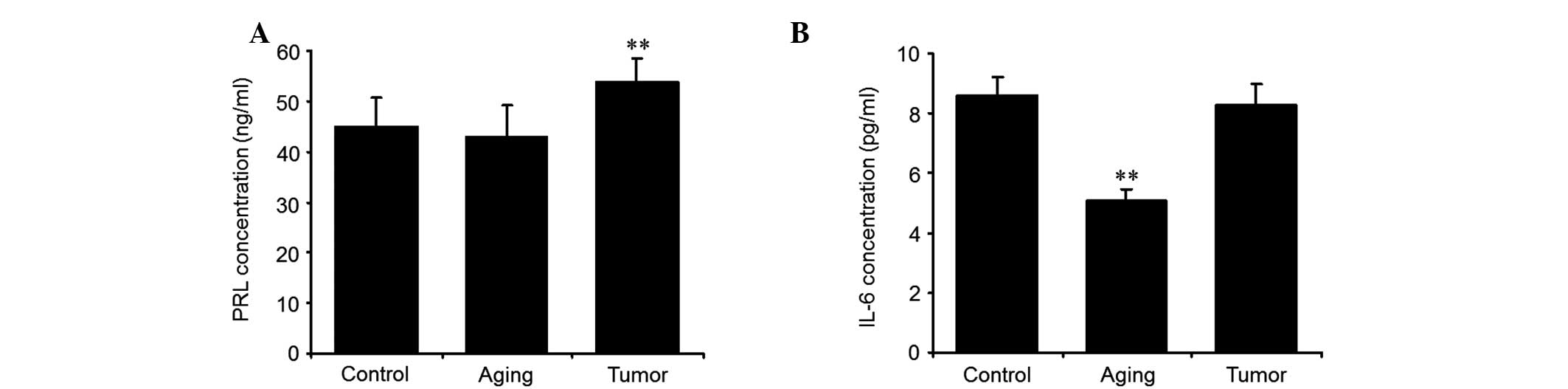
staining was also observed in this group (Fig. 2). The plasma concentration of PRL
was also detected using ELISA. The results revealed that the
concentration of plasma PRL was significantly increased in
DES-treated animals (Fig. 3A).
D-gal treatment of the rats in the aging group resulted in strong
senescence as evidenced by SA-β-Gal activity (Fig. 2). These results indicated that DES
implantation successfully induced the PRL adenoma in the rat
pituitary gland and that D-gal treatment promoted pituitary
aging.
Gene expression associated with cellular
senescence in aging and neoplasia pituitary gland
IL-6, one type of pleiotropic cytokine, exhibits
either inhibitory or stimulatory effects in several pituitary
tumors. IL-6 promotes pituitary proliferation in a paracrine manner
and inhibits the growth of the pituitary in an autocrine manner.
Plasma IL-6 was firstly detected using ELISA. The result revealed
that plasma IL-6 was lower in the aging group treated with D-gal
than in the control group. There was no significant difference
observed in the plasma IL-6 levels between the control and tumor
group (Fig. 3B). The expression
level of IL-6 in pituitary gland tissues was further detected using
qPCR. The expression of IL-6 was significantly increased in the
aging group compared with the control group. A significant decrease
in IL-6 expression was observed in the tumor group (Fig. 4A). The results indicated that IL-6
expression was increased in aging tissue but decreased in tumor
tissue. Upregulation of IL-6 in aging tissue and downregulation of
IL-6 in tumor tissue indicated that IL-6 expression was involved in
the pituitary aging process and tumor genesis. However, endogenous
IL-6 may underlie the slow proliferation rate and benign nature of
pituitary tumors. It is plausible that paracrine IL-6 effects may
allow initial pituitary cell growth, while autocrine IL-6 in the
same tumor triggers senescence and restrains aggressive growth and
malignant transformation.
The C/EBPβ transcription factor has been identified
as a critical gene that regulates PRL expression and activates the
expression of IL-6 (20). The
expression of C/EBPβ in pituitary tissue was also detected. The
results revealed that the expression of C/EBPβ was increased in the
aging group compared with the control group (Fig. 4B).
The Cdk inhibitor p21, as a major mediator of
cellular senescence, is one of the main targets of p53 for the
induction of cell cycle arrest in response to DNA damage. The qPCR
assay revealed that both p53 and p21 expression were significantly
increased in the aging group. A notable decrease in p21 expression
was observed in the tumor group (Fig.
4C and D). These results indicated that D-gal treatment
activated the expression of p53 and p21 which in turn contributed
to the senescence of pituitary tissue. Thus, D-gal may successfully
induce F344 rate to generate subacute senescent PRL adenoma.
The Cdk inhibitor p16 was also detected in pituitary
tissues. The expression of p16 was significantly increased in aging
pituitary tissue but was decreased in the pituitary tumor (Fig. 4E). Oncogene-induced senescence is
enabled via the p16/Rb or IL6/C/EBPβ senescence pathways. Our data
also suggest that the expression of p16 induces an age-dependent
decrease in the proliferative activity of pituitary cells and
unipotent progenitors.
The PTTG protein, initially isolated from pituitary
tumor cells, is abundantly expressed in pituitary tumors and
exhibits oncogene properties. The results revealed that the
expression of PTTG significantly decreased in the aging pituitary
and notably increased in pituitary tumors (Fig. 4F).
Discussion
In the present study we demonstrated that the
oncogene-induced senescence pathway IL-6/C/EBPβ and DNA damage
pathway p53/p21 as well as p16 were activated in the aging
pituitary gland treated by continuous injection of D-gal. No
significant activation of these pathways was observed in the
pituitary tumor induced by the DES implants. Pituitary tumor tissue
exhibited low expression of p21 and p16, both of which are cell
cycle suppressors. PPTG was downregulated in aging pituitary but
upregulated in pituitary tumors. Oncogene-induced senescence is
often accompanied by the upregulation of the CDK inhibitors
p16INK4A and p21, as well as by an increase in SA-β-Gal
activity.
D-gal, a reducing sugar, is normally present in the
body and is metabolized at normal concentration. When excessive
D-gal is exposed to an organism, it may be oxidized into aldehydes
and hydrogen peroxide, resulting in the accumulation of superoxide
anion and oxygen-derived free radicals (23,24).
This oxidative stress disturbs the mitochondrial function, destroys
cell structures and collapses the antioxidant mechanisms. Oxidative
stress and reactive oxygen species are the major causes of aging
(25). In the present study, we
have successfully induced the pituitary aging model using D-gal
treatment. Multiple cellular senescence pathways were activated
under oxidative stress caused by D-gal administration, including
the IL-6/C/EBPβ, p53/p21 and p16 pathways.
Continuous treatment of Fischer 344 rats with
estrogen results in the development of prolactin-secreting
pituitary tumors, which provides a well-defined animal model for
studying the pituitary adenomas (26). Commonly encountered, pituitary
adenomas are invariably benign and grow slowly. Premature
senescence is considered to be a significant mechanism in this
indolent growth (14,15). In the present study we detected the
expression of the proteins involved in premature senescence-related
pathways in pituitary tumors induced by DES implants. The results
demonstrated that the expression of p53 and C/EBPβ in pituitary
tumors was almost the same as that in the normal pituitary gland.
As essential tumor suppressors, the normal expression of p53 and
C/EBPβ may contribute to the limited proliferation of pituitary
tumors.
An increasing amount of evidence demonstrates that
cytokines play a significant role in the regulation of pituitary
physiology and that oncogene-induced senescence is linked
specifically to the activation of an inflammatory transcriptome.
The cytokine IL-6 has received special attention due to its
distinctive biological function. IL-6, detected in numerous cell
types, demonstrates roles in various biological processes including
immunity, inflammation, cancer and the aging process (20,27).
It has been reported that anterior pituitary cells express IL-6
(28). The production of IL-6 in
pituitary cells may be regulated by a number of compounds,
including IL-1 (28), estrogen
(29) and glucocorticoids
(30). IL-6 promotes pituitary
tumor growth but inhibits the proliferation of normal pituitary
cells (31). Our results revealed
that IL-6 expression was significantly upregulated in aging
pituitary tissue. This is consistent with a previous study which
revealed that IL-6 expression is accumulated in senescence cells
(20). These authors demonstrated
that IL-6 mediated oncogene-induced senescence in a cell-autonomous
mode. Its depletion caused the inflammatory network to collapse and
abolished senescence entry and maintenance. The expression of IL-6
is activated by C/EBPβ, and the results suggest that C/EBPβ and
IL-6 constitute a positive feedback network regulating
oncogene-induced senescence, and that C/EBPβ cooperates with IL-6
to amplify the activation of the inflammatory network (20). The expression of C/EBPβ was also
increased in the aging pituitary. However, the plasma IL-6
concentration was decreased in aging rats compared with normal
rats. This indicated that the paracrine activity of IL-6 was
inhibited in aging rats.
In the present study, the results revealed that the
expression of IL-6, C/EBPβ, p53, p21 and p16 was significantly
increased in aging pituitary tissues. Downregulation of IL-6, p21
and p16 was observed in pituitary tumors compared with normal
pituitary tissues. There was no significant change in the
expression of p53 and C/EBPβ in pituitary tumor cells compared with
normal ones. The expression of PTTG decreased in aging pituitary
tissues but increased in pituitary tumors. In conclusion, our
findings on the role of IL-6 in oncogene-induced senescence suggest
the involvement of endogenous IL-6 in the development of the
pituitary adenoma, and that pituitary cell growth regulation by
IL-6 underlies the role of cytokines as factors controlling
pituitary cell division. IL-6 in the same tumor triggers senescence
and restrains aggressive growth and malignant transformation.
Further experiments are required to assess the use of this pathway
as a target for effective therapy for tumor silencing and
prevention of adenoma progression towards malignancy.
Acknowledgements
This study was supported by Plans for Graduate
Research and Innovation in Colleges and Universities of Jiangsu
Province (cxzz12-0839) and the Science Foundation for Young
Scholars of Heilongjiang Province (QC2008C99).
References
|
1
|
Kovacs K, Horvath E and Vidal S:
Classification of pituitary adenomas. J Neurooncol. 54:121–127.
2001. View Article : Google Scholar
|
|
2
|
Melmed S: Mechanisms for pituitary
tumorigenesis: the plastic pituitary. J Clin Invest. 112:1603–1618.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ezzat S and Asa SL: Mechanisms of disease:
The pathogenesis of pituitary tumors. Nat Clin Pract Endocrinol
Metab. 2:220–230. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Levy A and Lightman S: Molecular defects
in the pathogenesis of pituitary tumours. Front Neuroendocrinol.
24:94–127. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Melmed S: Pathogenesis of pituitary
tumors. Nat Rev Endocrinol. 7:257–266. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wierinckx A, Raverot G, Nazaret N,
Jouanneau E, Auger C, Lachuer J, et al: Proliferation markers of
human pituitary tumors: contribution of a genome-wide transcriptome
approach. Mol Cell Endocrinol. 326:30–39. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Scheithauer BW, Gaffey TA, Lloyd RV, Sebo
TJ, Kovacs KT, Horvath E, et al: Pathobiology of pituitary adenomas
and carcinomas. Neurosurgery. 59:341–353. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Arzt E, Chesnokova V, Stalla GK and Melmed
S: Pituitary adenoma growth: a model for cellular senescence and
cytokine action. Cell Cycle. 8:677–678. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Herbig U, Jobling WA, Chen BP, Chen DJ and
Sedivy JM: Telomere shortening triggers senescence of human cells
through a pathway involving ATM, p53, and p21(CIP1), but not
p16(INK4a). Mol Cell. 14:501–513. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
d’Adda di Fagagna F, Reaper PM,
Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, et al: A DNA
damage checkpoint response in telomere-initiated senescence.
Nature. 426:194–198. 2003. View Article : Google Scholar
|
|
11
|
Takai H, Smogorzewska A and de Lange T:
DNA damage foci at dysfunctional telomeres. Curr Biol.
13:1549–1556. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Serrano M and Blasco MA: Putting the
stress on senescence. Curr Opin Cell Biol. 13:748–753. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chesnokova V, Zonis S, Rubinek T, Yu R,
Ben-Shlomo A, Kovacs K, et al: Senescence mediates pituitary
hypoplasia and restrains pituitary tumor growth. Cancer Res.
67:10564–10572. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chesnokova V, Zonis S, Kovacs K,
Ben-Shlomo A, Wawrowsky K, Bannykh S, et al: p21(Cip1) restrains
pituitary tumor growth. Proc Natl Acad Sci USA. 105:17498–17503.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chesnokova V, Zhou C, Ben-Shlomo A, Zonis
S, Tani Y, Ren SG, et al: Growth hormone is a cellular senescence
target in pituitary and nonpituitary cells. Proc Natl Acad Sci USA.
110:E3331–E3339. PubMed/NCBI
|
|
16
|
Collado M, Gil J, Efeyan A, Guerra C,
Schuhmacher AJ, Barradas M, et al: Tumour biology: senescence in
premalignant tumours. Nature. 436:6422005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mooi WJ and Peeper DS: Oncogene-induced
cell senescence-halting on the road to cancer. N Engl J Med.
355:1037–1046. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kuilman T and Peeper DS:
Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev
Cancer. 9:81–94. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Collado M, Blasco MA and Serrano M:
Cellular senescence in cancer and aging. Cell. 130:223–233. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kuilman T, Michaloglou C, Vredeveld LC,
Douma S, van Doorn R, Desmet CJ, et al: Oncogene-induced senescence
relayed by an interleukin-dependent inflammatory network. Cell.
133:1019–1031. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Abbud RA, Takumi I, Barker EM, Ren SG,
Chen DY, Wawrowsky K, et al: Early multipotential pituitary focal
hyperplasia in the alpha-subunit of glycoprotein hormone-driven
pituitary tumor-transforming gene transgenic mice. Mol Endocrinol.
19:1383–1391. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lloyd RV: Estrogen-induced hyperplasia and
neoplasia in the rat anterior pituitary gland. An
immunohistochemical study. Am J Pathol. 113:198–206.
1983.PubMed/NCBI
|
|
23
|
Lu J, Zheng YL, Luo L, Wu DM, Sun DX and
Feng YJ: Quercetin reverses D-galactose induced neurotoxicity in
mouse brain. Behav Brain Res. 171:251–260. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wu DM, Lu J, Zheng YL, Zhou Z, Shan Q and
Ma DF: Purple sweet potato color repairs d-galactose-induced
spatial learning and memory impairment by regulating the expression
of synaptic proteins. Neurobiol Learn Mem. 90:19–27. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Olanow CW: A radical hypothesis for
neurodegeneration. Trends Neurosci. 16:439–444. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Spady TJ, McComb RD and Shull JD: Estrogen
action in the regulation of cell proliferation, cell survival, and
tumorigenesis in the rat anterior pituitary gland. Endocrine.
11:217–233. 1999. View Article : Google Scholar
|
|
27
|
Naugler WE and Karin M: The wolf in
sheep’s clothing: the role of interleukin-6 in immunity,
inflammation and cancer. Trends Mol Med. 14:109–119. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Spangelo BL, Judd AM, Isakson PC and
MacLeod RM: Interleukin-1 stimulates interleukin-6 release from rat
anterior pituitary cells in vitro. Endocrinology. 128:2685–2692.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nagashima AC, Giacomini D, Castro CP,
Pereda MP, Renner U, Stalla GK, et al: Transcriptional regulation
of interleukin-6 in pituitary folliculo-stellate TtT/GF cells. Mol
Cell Endocrinol. 201:47–56. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Schöbitz B, Van Den Dobbelsteen M,
Holsboer F, Sutanto W and De Kloet ER: Regulation of interleukin 6
gene expression in rat. Endocrinology. 132:1569–1576.
1993.PubMed/NCBI
|
|
31
|
Arzt E, Buric R, Stelzer G, Stalla J,
Sauer J, Renner U, et al: Interleukin involvement in anterior
pituitary cell growth regulation: effects of IL-2 and IL-6.
Endocrinology. 132:459–467. 1993.PubMed/NCBI
|