Introduction
Tuberculosis is a common infectious disease usually
caused by Mycobacterium tuberculosis (M.tb) (1). Tuberculosis most regularly affects
the lungs, but can potentially affect almost any other body organ.
M.tb is spread when individuals with active M.tb infection cough,
sneeze, or otherwise transmit respiratory fluids through the air
(2). The majority of M.tb
infections are asymptomatic and latent, but approximately one in
ten latent infections eventually progresses to active tuberculosis
which, if left untreated, is fatal in >50% of cases.
Peroxisome proliferator-activated receptor (PPAR), a
member of the lipid-activated nuclear receptor family, is a key
transcriptional regulator of cell differentiation, inflammation,
and lipid metabolism in macrophages and dendritic cells (3). PPARs are expressed in leukocytes,
including macrophages, dendritic cells, T cells and B cells, and a
role for these receptors in inflammation and immunoregulation has
previously been proposed (4,5).
There are three types of human PPARs: α, γ, and β/δ, and each type
is the product of a different gene (6). PPARγ, a nuclear receptor superfamily
member, is a transcriptional factor that regulates inflammation and
is highly expressed in alternatively activated alveolar macrophages
and macrophage-derived foam cells, both of which are closely
associated with the pathogenesis of tuberculosis (7). A previous study demonstrated that
PPARγ is involved in lipid body biogenesis, revealing a cross-talk
between the innate immune receptor Toll-like receptor 2 (TLR2) and
the lipid-activated nuclear receptor PPARγ that coordinates lipid
metabolism and inflammation in the Bacillus Calmette-Guérin
(BCG)-infected macrophages, thereby potentially altering
mycobacterial pathogenesis (8).
Although it is well established that PPARγ acts as a master
regulator in lipid metabolism and inflammation, the involvement of
PPARγ in the immune response of macrophages to intracellular
pathogen infection remains to be elucidated.
Tumor necrosis factor α (TNF-α) is crucial in
establishing and maintaining the inflammatory response against
infections (9). The blockage of
TNF-α has marked effects on the progression of tuberculosis in
experimental models. For example, neutralization of TNF-α in a
murine model has been demonstrated to lead to tuberculosis
aggravation or reactivation (10).
Furthermore, increased levels of TNF-α are frequently detected in
the culture supernatants of peripheral blood mononucleated cells
from patients with pulmonary tuberculosis stimulated with
mycobacterial antigens (11,12).
Similar to TNF-α, IL-6 is involved in chronic inflammatory diseases
(13). IL-6-deficient
(IL-6−/−) mice are resistant to the induction of various
experimental inflammatory diseases (14). The mitogen-activated protein kinase
(MAPK) pathway is important for immune response and mycobacterial
pathogenesis (15–17), and MAPK family members include
extracellular signal-regulated kinase, p38 MAPK (p38), and
stress-activated protein kinase/c-Jun N-terminal kinase (18). It has been reported that p38 was
activated in monocytes following M.tb infection (19). The involvement of TLR2 in the M.tb
infection is well-defined, as patients with TLR2 polymorphisms
exhibit increased susceptibility to M.tb infection, whereas
TLR2−/− mice are unable to mount optimal immune
responses against mycobacteria (20). However, potential additional
signaling pathways involved in M.tb-induced molecular regulation
are unknown.
The aim of the current study was to investigate the
role of PPARγ in P19-induced immune responses, including TLR2
activation, p38 phosphorylation and cytokine production.
Materials and methods
Reagents and antibodies
PPARγ agonist BRL49653, antagonist GW-9662 and fetal
bovine serum (FBS) were purchased from Cayman Chemical Co. (Ann
Arbor, MI, USA). Cell culture reagents, medium, L-glutamine and
antibiotics were obtained from Gibco-BRL (Rockville, MD, USA).
Monoclonal antibodies (all produced from immunized rabbits) against
PPARγ, phospho-p38, MAPK (Thr180/Tyr182), total p38, TLR2 and GAPDH
were purchased from Cell Signaling Technology, Inc. (Danvers, MA,
USA). TLR2 small interfering (si)RNA and control siRNA (fluorescein
isothiocyanate-conjugated) were purchased from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA).
Bacterial strains and P19 isolation
Lyophilized M.tb H37Rv (ATCC 25618) and
Mycobacterium smegmatis (M. smegmatis; ATCC 700084)
were obtained from American Type Culture Collection (ATCC;
Manassas, VA, USA), reconstituted and used as described previously
(21). The concentration of
bacteria was determined by counting in a Petroff-Hausser chamber
(Hede Biotechnology, Beijing, China). Bacteria prepared in this
manner are ≥90% viable as assessed by colony forming unit
assays.
Purified M.tb 19-kDa lipoprotein (P19) was obtained
as described previously (22). In
brief, cell-wall fractions were obtained by sonication of suspended
M.tb H37Rv at 20 kHz in iced water (5 cycles for 5 min each).
Protein (40 μg) were mixed with a reducing sample buffer (0.05 mM
EDTA, 0.1% SDS, 1% glycerol, 10% 2-mercaptoethanol, and 0.5 mM/ml
Tris-HCl pH 6.8), heated for 5 min at 95°C and loaded onto 12%
SDS-PAGE gels. Following electrophoresis, proteins were transferred
to a polyvinylidene fluoride (PVDF) membrane and stained with
Ponceau S red (Energy Chemicals, Shanghai, China) to identify the
19 kDa band; the identity of this band was confirmed in parallel
blots with the IT-19 monoclonal antibody. The band was then
excised, solubilized in dimethylsulfoxide (DMSO) and precipitated
with carbonate/bicarbonate sodium buffer (0.05 M, pH 9.6). The
pellet was rinsed three times with phosphate-buffered saline (PBS;
pH 7.4) and stored at −20°C. The concentration of the protein was
measured with the Bradford Protein Assay kit (Bio-Rad, Hercules,
CA, USA).
Cell culture and mycobacterial
infection
The WBC 264-9C macrophage cell line (HB-8902; ATCC)
was cultivated in cultured in RPMI-1640 medium supplemented with
15% FBS, 10 mM HEPES [4-(2-hydroxyethyl)-1-piperazineethanesulfonic
acid], 2 mM L-glutamine, and 50 μg/ml gentamicin at 37°C in a
humidified incubator containing 5% CO2. For P19
stimulation or infection with M.tb H37Rv or M. smegmatis,
WBC 264-9C cells were placed in 12-well culture plates with glass
coverslips at a density of 5×105 cells/ml for 24 h.
Cells were then washed and incubated for an additional 18 h in the
medium with 0.1% FBS. The macrophage monolayers in the tissue
culture plates were washed with prewarmed RPMI-1640 medium and
replaced with 1 ml RPMI-1640 medium containing 10 mM HEPES and 0.4%
human serum albumin, and viable M.tb H37Rv or M. smegmatis
at a multiplicity of infection of 1:10 macrophage/bacteria, or
treated with 5 μg/ml P19, and incubated at 37°C for various time
periods prior to analysis. Alternatively, cells were pretreated
with BRL49653 (5 μM), GW-9662 (1 μM) or 0.01% DMSO (as
vehicle-control) at 37°C for 30 min prior to bacterial infection.
The cells and cell-free supernatants were collected and stored at
−20°C for subsequent assays. Cell viability was assessed by a
trypan blue (Energy Chemicals) exclusion method at the end of each
experiment (viability of ≥90% was required).
TLR2 siRNA transfection
To silence the expression of TLR2, macrophages were
transfected with the TLR2 siRNA. The siRNA transfection was
performed according to the manufacturer’s instructions. In brief,
WBC 264-9C macrophages were incubated in the siRNA Transfection
medium (Santa Cruz Biotechnology, Inc.) at a density of
2×105 cells/well in 12-well cell culture plates,
followed by the addition of the TLR2 siRNA or negative control
siRNA, and incubated at room temperature for 30 min. A transfection
efficiency of >95% was demonstrated by flow cytometry (FACS
Calibur, Becton Dickinson, Franklin Lakes, NJ, USA). Following
transfection, the TLR2 expression was analyzed by reverse
transcription (RT)-quantitative polymerase chain reaction (qPCR)
and western blot analysis. The cytokine expression levels in the
supernatant of the transfected macrophages were measured by
ELISA.
Western blot analysis
The macrophages were rinsed with prewarmed PBS, and
lysed in an ice-cold extraction buffer (50 mM Tris, pH 7.5; 150 mM
NaCl; 10% glycerol; 1 mM EDTA; 1 mM EGTA; 1% NP-40; 1 mM
dithiothreitol; and protease inhibitor cocktail (Roche, Basel,
Switzerland). The homogenate was incubated on ice for 20 min, then
centrifuged at 13,000 × g for 20 min at 4°C. The supernatant was
collected, and the concentration of the protein in the supernatant
was determined using the Bradford Protein Assay kit. The whole cell
lysates from the macrophages were subjected to 12% SDS-PAGE, and
subsequently blotted onto a PVDF membrane. The membrane was
incubated with the antibodies of interest. GAPDH was used as an
internal control. The quantitation of protein bands was performed
using the Quantity One software (Bio-Rad, Hercules, CA, USA).
RT-qPCR
Total RNA from the macrophages was extracted using
an RNAiso Plus kit (Takara, Dalian, China) according to the
manufacturer’s instructions. The extracted total RNA was quantified
by absorbance at 280 nm using a NanoDrop 2000c Spectrophotometer
(Thermo Fisher Scientific, Waltham, MA, USA). The mRNA in the total
RNA was reverse transcribed to complementary DNA using a
PrimeScript RT Reagents kit (Takara). qPCR was performed in the
iCycler iQ5 (Bio-Rad) using SYBR Premix Ex Taq II (Takara) with the
following conditions: 30 sec at 95°C, 40 cycles of 5 sec at 95°C,
and 30 sec at 60°C. The mRNA expression levels, which were
normalized against GAPDH, were calculated and expressed as
2−ΔΔCT. The primers used for qPCR were as follows:
PPARγ: F 5′-CATTCTGGCCCACCAACTTTGG-3′, and R 5′-TGG
AGATGCAGGCTCCACTTTG-3′ (229 bp); TRL2: F 5′-AAG
AGGAAGCCCAAGAAAGC-3′, and R 5′-CAATGG GAATCCTGCTCACT-3′ (80 bp);
GAPDH: F 5′-ATGGGG AAGGTGAAGGTCG-3′, and R 5′-GGGGTCATT
GATGGCAACAA-3′ (156 bp).
Cytokine assays
Following infection with M.tb H37Rv or M.
smegmatis, or treatment with P19, the levels of the cytokines
(IL-6 and TNF-α) from the supernatant of the macrophages were
measured with ELISA kits (R&D Systems, Minneapolis, MN, USA)
according to the manufacturer’s instructions.
Statistical analysis
All data in the present study were analyzed by the
statistics package SPSS, version 13.0 (SPSS, Inc., Chicago, IL,
USA). All data are expressed as the mean ± standard error. A direct
comparison between two groups was conducted with the Student’s
non-paired t-test, and analysis of variance (ANOVA) with Tukey’s
post-hoc tests was used to compare the means of three or more
groups. P<0.05 was considered to represent a statistically
significant difference.
Results
PPARγ expression, p38 phosphorylation and
cytokine production were upregulated following M.tb H37Rv infection
or P19 treatment
To investigate the effect of P19 treatment on PPARγ
expression, p38 phosphorylation and cytokine production, human
macrophages were incubated with 5 μg/ml purified P19 for various
time periods (from 0 to 48 h). M.tb H37Rv or M. smegmatis
infection were used as controls. The expression of PPARγ was
significantly upregulated by M.tb H37Rv infection (F(4,10)=35.34, P<0.0001) and P19
stimulation (F(4,10)=19.24, P<0.01) (Fig. 1A). Post hoc analysis revealed that
the expression levels of PPARγ were upregulated at 48 h compared
with the levels at 0 h, following M.tb infection (5.40±0.40-fold,
P<0.0001) or P19 treatment (4.10±0.41-fold, P<0.0001). M.
smegmatis infection did not significantly affect PPARγ levels
(F(4,10)=0.11, P=0.87), as expected. The
phosphorylation level of p38 was expressed as the ratio of
phospho-p38 to total p38 (Fig.
1B). ANOVA revealed significant differences in levels of p38
phosphorylation following M.tb H37Rv infection (F(4,10)=35.34, P<0.0001) and
following P19 treatment (F(4,10)=19.24, P<0.01). Phospho-p38
levels were 3.11±0.20-fold (P<0.01, M.tb H37Rv infection) and
2.50±0.34-fold (P<0.01, P19 treatment) greater at 48 h than
those at 0 h, whereas p38 phosphorylation was not significantly
affected by M. smegmatis infection (F(4,10)=0.11, P=0.87). The levels of
IL-6 (Fig. 1C) and TNF-α (Fig. 1D) were also analyzed. Two-way ANOVA
revealed significant effects of time (Ftime(4,30)=16.29,
P<0.0001) and infection/treatment
(Finfection(2,30)=57.64, P<0.0001) on IL-6
production, as well as an interaction between them
(Finteraction(8,30)=4.46, P<0.01). Similarly, the
levels of TNF-α were also significantly affected by time
(Ftime(4,30)=25.92, P<0.0001) and infection/treatment
(Finfection(2,30)=3.94, P<0.05); however, no
significant effect of time-infection interaction was detected
(Finteraction(8,30)=2.95, P=0.07).
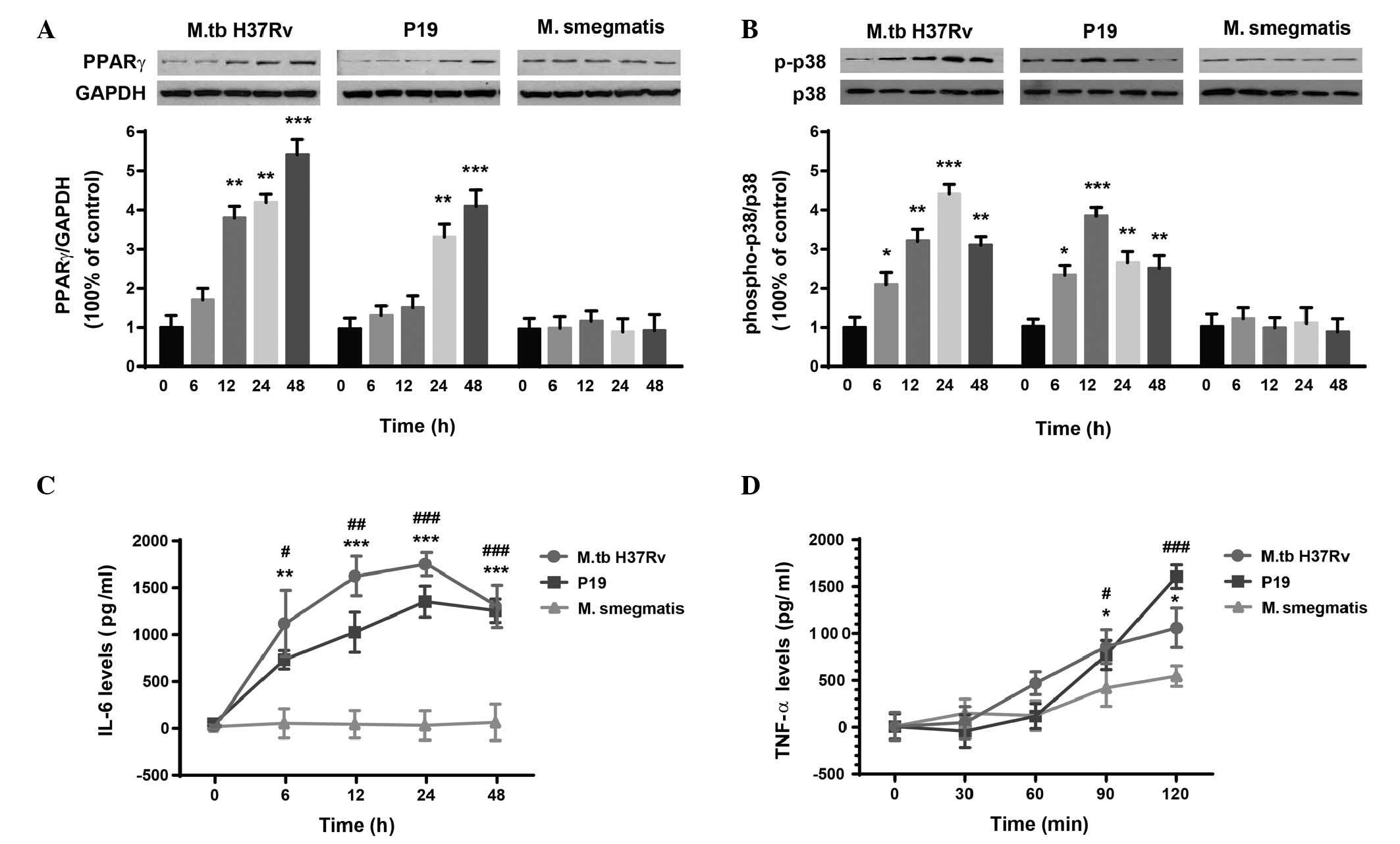 | Figure 1M.tb H37Rv and P19 upregulate the
expression of (A) PPARγ, (B) p38 phosphorylation, (C) IL-6 and (D)
TNF-α in human macrophages. As a control, macrophages were also
incubated with M. smegmatis. The phosphorylation state of
p38 was expressed as the ratio of phospho-p38 to total p38. Data
are expressed as the mean ± standard error (n=3). For (A) and (B):
*P<0.05, **P<0.01,
***P<0.0001 vs. 0 h within each group. For (C) and
(D): *P<0.05, **P<0.01,
***P<0.0001, M.tb H37Rv infection vs. M.
smegmatis infection at the same time point;
#P<0.05, ##P<0.01,
###P<0.0001, P19 treatment vs. M. smegmatis
infection at the same time point. PPAR, peroxisome
proliferator-activated receptor; M.tb, Mycobacterium
tuberculosis; P19, M.tb 19 kDa lipoprotein; M.
smegmatis, Mycobacterium smegmatis; TNF, tumor necrosis
factor. |
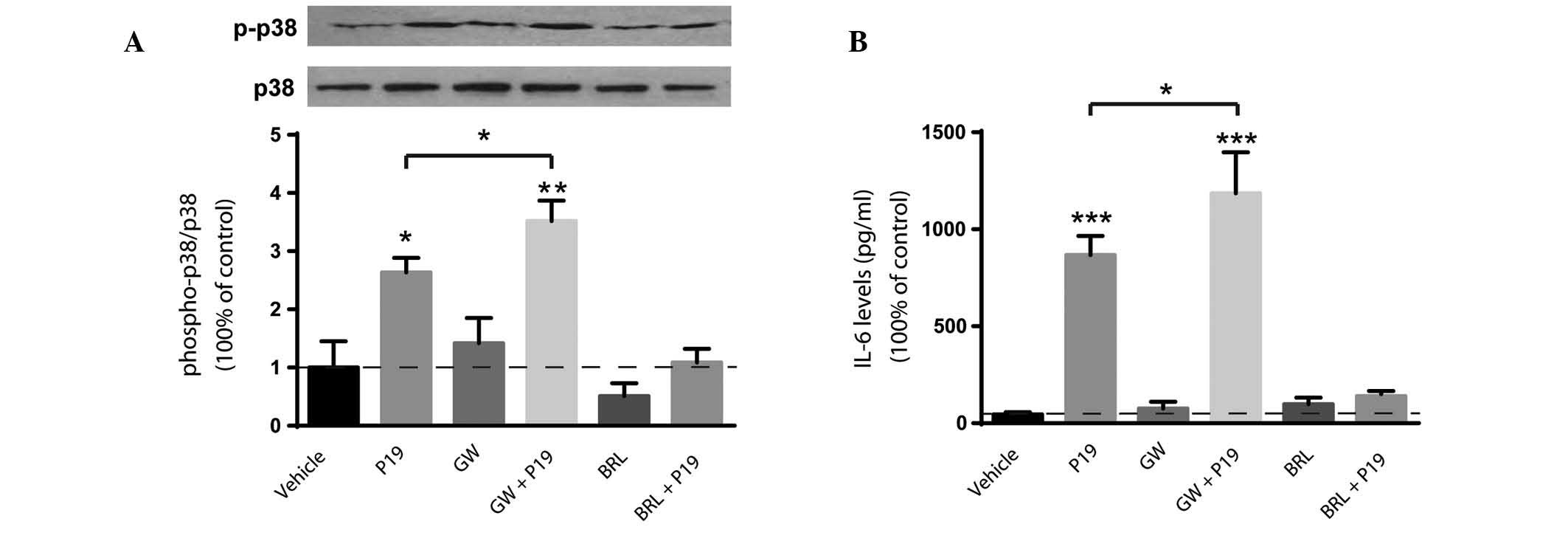
P19-induced p38 phosphorylation and IL-6
production is modulated by PPARγ
Since M.tb H37Rv infection and P19 treatment
upregulate the expression of PPARγ, the involvement of PPARγ in p38
phosphorylation and cytokine production induced by P19 in
macrophages was investigated. As expected, the levels of p38
phosphorylation were significantly elevated following 24-h P19
treatment compared with those in the vehicle control (P<0.05;
Fig. 2A). Pretreatment with a
specific PPARγ agonist (BRL49653) or antagonist (GW-9662) did not
significantly affect p38 phosphorylation compared with that in the
vehicle control. P19 treatment combined with GW-9662 pretreatment
significantly upregulated p38 phosphorylation (P<0.01 vs.
vehicle control). Additionally, the activation of PPARγ by BRL49653
attenuated P19-induced upregulation of p38 phosphorylation
(P=0.21).
Similarly, 24 h after P19 treatment, the expression
levels of IL-6 were significantly elevated (P<0.0001 vs.
control; Fig. 2B). The expression
levels of IL-6 were not significantly affected by treatment with
the PPAR agonist or antagonist alone. However, when compared with
P19 alone, treatment with the PPARγ antagonist significantly
upregulated the levels of IL-6 production (P<0.05), while the
PPARγ agonist attenuated them (P=0.72).
P19-induced PPARγ expression, p38
phosphorylation and cytokine production are dependent on TLR2
To confirm the involvement of the TLR2 in
P19-induced PPARγ expression and p38 phosphorylation, the
expression of PPARγ, p38, IL-6 and TNF-α were measured in TLR2
knockdown macrophages (Fig. 3A).
Two-way ANOVA revealed a significant effect of TLR2 knockdown
(FTLR2-siRNA(1,8)=70.4, P<0.0001) and P19
treatment (FP19(1,8)=23.3, P<0.01) on PPARγ mRNA
expression, as well as an interaction between them
(Finteraction(1,8)=21.8, P<0.01) (Fig. 3B). Tukey’s post hoc tests indicated
that the levels of PPARγ mRNA were significantly reduced following
TLR2 knockdown, regardless of whether P19 treatment is used or not
(P<0.0001 vs. control).
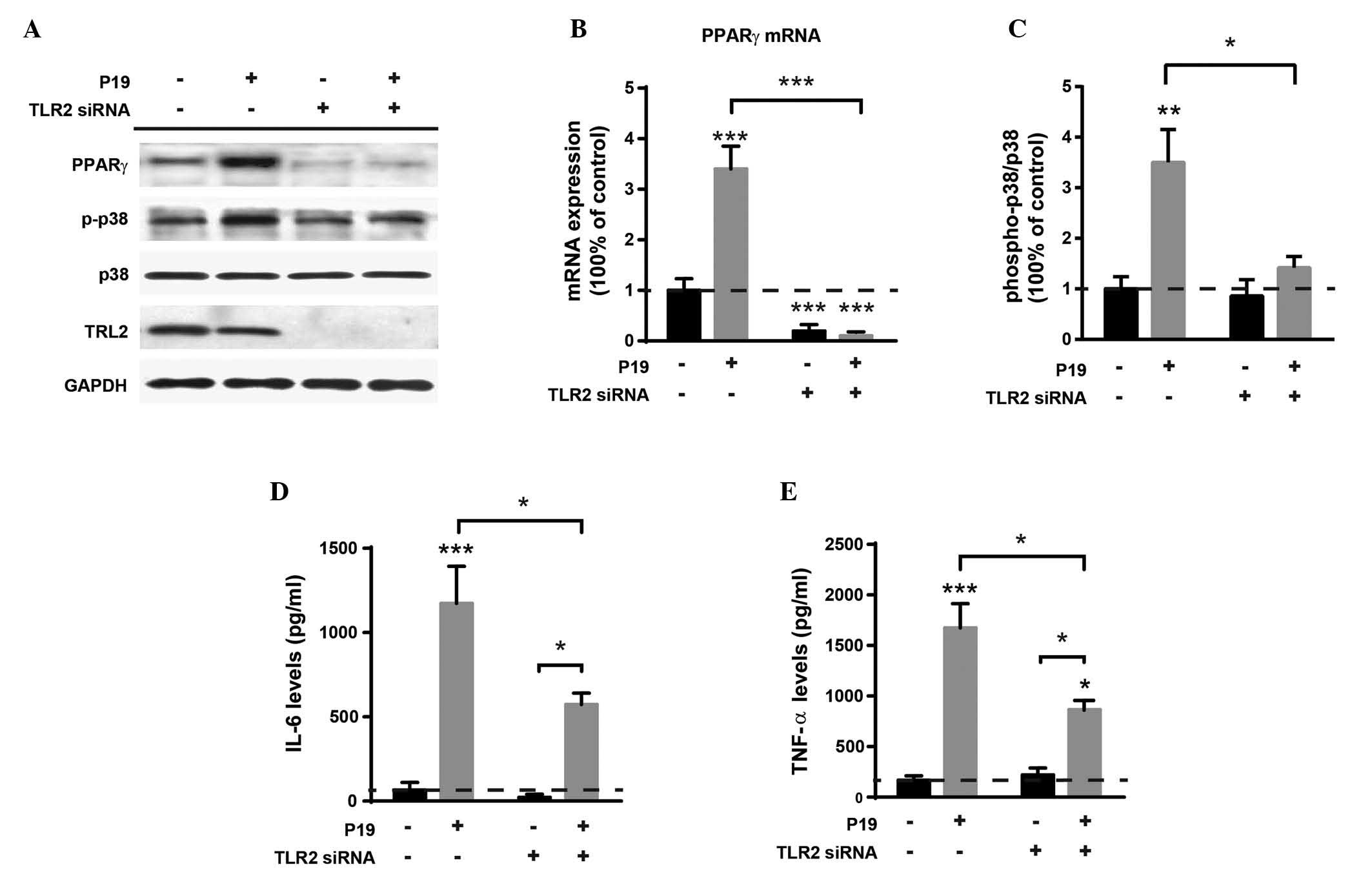 | Figure 3P19-induced PPARγ expression, p38
phosphorylation and cytokine production in macrophages are
dependent on TLR2. (A) A representative western blot of PPARγ,
phospho-p38 and total p38 expression. The expression levels of TLR2
following TLR2 knockdown by TLR2 siRNA were also determined. GAPDH
was used as a loading control. (B) PPARγ mRNA expression. (C) The
levels of p38 phosphorylation, expressed as the ratio of
phospho-p38 to total p38. (D) The levels of IL-6. (E) The levels of
TNF-α. Data are expressed as the mean ± standard error (n=3).
*P<0.05, **P<0.01,
***P<0.0001 vs. the control unless otherwise
indicated. TLR, Toll-like receptor; P19, M.tb 19kDa lipoprotein;
PPAR, peroxisome proliferator-activated receptor; si, small
interfering; TNF, tumor necrosis factor. |
With regards to p38 phosphorylation, significant
effects of TLR2 knockdown (FTLR2-siRNA(1,8)=7.81, P<0.05), P19 treatment
(FP19(1,8)=14.8, P<0.01) and interaction
factor (Finteraction(1,8)=5.97, P<0.05) were also
detected (Fig. 3C). As expected,
the levels of phospho-p38 were increased following P19 treatment
(P<0.01 vs. control). In the absence of P19 treatment, no
significant difference in the levels of p38 phosphorylation between
the TLR2 knockdown macrophages and negative siRNA-treated
macrophages was observed (P=0.94). However, TLR2 knockdown combined
with P19 treatment significantly decreased the levels of p38
phosphorylation (P<0.01), compared with levels following P19
treatment without TLR2 knockdown. The levels of IL-6 and TNF-α were
also analyzed (Fig. 3D and E,
Table I). The results demonstrated
that TLR2 knockdown combined with P19 treatment significantly
reduced the expression levels of both IL-6 and TNF-α compared with
the levels following P19 treatment without TLR2 knockdown.
 | Table IStatistical results for IL-6 and TNF-α
production. |
Table I
Statistical results for IL-6 and TNF-α
production.
| Factor | F(1,8)
value | P-value |
|---|
| IL-6 levels |
| P19 | 49.80 | <0.0001c |
| TLR2-siRNA | 7.48 | 0.0256a |
| Interaction | 5.62 | 0.0453a |
| TNF-α levels |
| P19 | 62.80 | <0.0001c |
| TLR2-siRNA | 7.74 | 0.0239a |
| Interaction | 10.10 | 0.0131a |
Discussion
Modulation of the host immune response is essential
in mycobacterial pathogenesis. M.tb enhances its survival in
macrophages by suppressing immune responses, in part through its
complex cell wall structures. The purified mycobacterial cell wall
lipoprotein P19 is well-defined as a TLR2 agonist (23). PPARγ is a prime candidate for an
intracellular molecular switch based on its central role in
controlling the inflammatory response in macrophages (24), and although PPARγ has been
extensively investigated in other diseases (25), its immunoregulatory role in
infectious diseases (particularly tuberculosis) is just beginning
to be recognized (8,26,27).
In the present study, the expression of PPARγ in human macrophages
was enhanced by M.tb H37Rv and P19, but not M. smegmatis. It
was also observed that P19 strongly induces p38 phosphorylation and
cytokine (IL-6 and TNF-α) production.
A previous study established that the activation of
PPARγ may repress target inflammatory genes, including
proinflammatory cytokines and inducible nitric oxide synthase,
through ligand-dependent transrepression of NF-κB target genes
(28). de Assis et al
(29) demonstrated that the PPARγ
agonist BRL49653 potentiates lipid body biogenesis in peritoneal
macrophages following oxidized phospholipid stimulation. A similar
role for PPARγ in lipid body biogenesis was reported in M.
bovis BCG, but not M. smegmatis (8). In the present study, the effect of
PPARγ activation during P19 infection was analyzed. It was
demonstrated that pretreatment with the PPARγ antagonist GW-9662
significantly upregulated P19-induced p38 phosphorylation and IL-6
production. However, pretreatment with the PPARγ agonist BRL49653
or antagonist GW-9662 alone did not affect p38 phosphorylation
compared with that of the vehicle control. These results indicate
that PPARγ may modulate P19-induced immune response through p38
phosphorylation.
Since TLRs and PPARγ contribute to M.tb-induced
immune responses and have been indicated to regulate host
susceptibility to pathogens, TLR2 activation involvement in the
regulation of PPARγ expression, p38 phosphorylation and cytokine
production in the presence of P19 treatment was investigated in the
current study. Following TLR2 knockdown in macrophages, the
expression of PPARγ was significantly decreased in the presence or
absence of P19 treatment. Furthermore, the levels of p38
phosphorylation and cytokine production were significantly reduced
in TLR2 knockdown macrophages in the presence of P19 treatment
compared with those in non-knockdown macrophages treated with P19.
These results demonstrated that PPARγ expression, p38
phosphorylation and cytokine production in human macrophages are
associated with TLR2. Results of the present study are consistent
with those of previous studies that observed Mycobacterium
bovis BCG-induced PPARγ expression, lipid body formation, and
PGE2 generation inhibition in TLR2-deficient mice
(8).
In conclusion, the findings of the current study
demonstrate that the M.tb cell wall component P19 induces PPARγ
expression in a TLR2-dependent manner. In the TLR2-dependent
signaling pathway, PPARγ acts as a key modulator of inflammation in
P19-stimulated macrophages. These findings suggest a role for PPARγ
and TLR2 in P19-induced p38 phosphorylation and cytokine
production, thereby potentially affecting the M.tb pathogenesis.
Future studies in animal models are required to further
characterize the role of PPARγ and TLR2 in the pathogenesis of
tuberculosis.
References
|
1
|
Kumar V, Abbas AK, Fausto N and Mitchell
RN: Robbins Basic Pathology. 8th edition. Saunders Elsevier;
Philadelphia, PA: pp. 516–522. 2007
|
|
2
|
Konstantinos A: Testing for tuberculosis.
Aust Prescr. 33:12–18. 2010.
|
|
3
|
Szatmari I and Nagy L: Nuclear receptor
signalling in dendritic cells connects lipids, the genome and
immune function. EMBO J. 27:2353–2362. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Szanto A and Nagy L: Retinoids potentiate
peroxisome proliferator-activated receptor gamma action in
differentiation, gene expression, and lipid metabolic processes in
developing myeloid cells. Mol Pharmacol. 67:1935–1943. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Szatmari I, Töröcsik D, Agostini M, et al:
PPARgamma regulates the function of human dendritic cells primarily
by altering lipid metabolism. Blood. 110:3271–3280. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Braissant O, Foufelle F, Scotto C, Dauça M
and Wahli W: Differential expression of peroxisome
proliferator-activated receptors (PPARs): tissue distribution of
PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology.
137:354–366. 1996.PubMed/NCBI
|
|
7
|
Rigamonti E, Fontaine C, Lefebvre B, et
al: Induction of CXCR2 receptor by peroxisome
proliferator-activated receptor gamma in human macrophages.
Arterioscler Thromb Vasc Biol. 28:932–939. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Almeida PE, Silva AR, Maya-Monteiro CM, et
al: Mycobacterium bovis bacillus Calmette-Guérin infection induces
TLR2-dependent peroxisome proliferator-activated receptor gamma
expression and activation: functions in inflammation, lipid
metabolism, and pathogenesis. J Immunol. 183:1337–1345. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bruns H, Meinken C, Schauenberg P, et al:
Anti-TNF immunotherapy reduces CD8+ T cell-mediated
antimicrobial activity against Mycobacterium tuberculosis in
humans. J Clin Invest. 119:1167–1177. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mohan VP, Scanga CA, Yu K, et al: Effects
of tumor necrosis factor alpha on host immune response in chronic
persistent tuberculosis: possible role for limiting pathology.
Infect Immun. 69:1847–1855. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dlugovitzky D, Bay ML, Rateni L, et al:
Influence of disease severity on nitrite and cytokine production by
peripheral blood mononuclear cells (PBMC) from patients with
pulmonary tuberculosis (TB). Clin Exp Immunol. 122:343–349. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Al-Attiyah R, El-Shazly A and Mustafa AS:
Comparative analysis of spontaneous and mycobacterial
antigen-induced secretion of Th1, Th2 and pro-inflammatory
cytokines by peripheral blood mononuclear cells of tuberculosis
patients. Scand J Immunol. 75:623–632. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jones SA, Scheller J and Rose-John S:
Therapeutic strategies for the clinical blockade of IL-6/gp130
signaling. J Clin Invest. 121:3375–3383. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Silva LC, Ortigosa LC and Benard G:
Anti-TNF-α agents in the treatment of immune-mediated inflammatory
diseases: mechanisms of action and pitfalls. Immunotherapy.
2:817–833. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Roach SK and Schorey JS: Differential
regulation of the mitogen-activated protein kinases by pathogenic
and nonpathogenic mycobacteria. Infect Immun. 70:3040–3052. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Senthil KA, Bansal K, Holla S, Verma-Kumar
S, Sharma P and Balaji KN: ESAT-6 induced COX-2 expression involves
coordinated interplay between PI3K and MAPK signaling. Mol Immunol.
49:655–663. 2012. View Article : Google Scholar
|
|
17
|
Lee SH, Kim DW, Back SS, et al: Transduced
Tat-Annexin protein suppresses inflammation-associated gene
expression in lipopolysaccharide (LPS)-stimulated Raw 264.7 cells.
BMB Rep. 44:484–489. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang W and Liu HT: MAPK signal pathways
in the regulation of cell proliferation in mammalian cells. Cell
Res. 12:9–18. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Surewicz K, Aung H, Kanost RA, Jones L,
Hejal R and Toossi Z: The differential interaction of p38 MAP
kinase and tumor necrosis factor-alpha in human alveolar
macrophages and monocytes induced by Mycobacterium tuberculois.
Cell Immunol. 228:34–41. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Andersson M, Lutay N, Hallgren O,
Westergren-Thorsson G, Svensson M and Godaly G: Mycobacterium bovis
bacilli Calmette-Guerin regulates leukocyte recruitment by
modulating alveolar inflammatory responses. Innate Immun.
18:531–540. 2012. View Article : Google Scholar
|
|
21
|
Schlesinger LS, Bellinger-Kawahara CG,
Payne NR and Horwitz MA: Phagocytosis of Mycobacterium tuberculosis
is mediated by human monocyte complement receptors and complement
component C3. J Immunol. 144:2771–2780. 1990.PubMed/NCBI
|
|
22
|
Sánchez A, Espinosa P, García T and
Mancilla R: The 19 kDa Mycobacterium tuberculosis lipoprotein
(LpqH) induces macrophage apoptosis through extrinsic and intrinsic
pathways: a role for the mitochondrial apoptosis-inducing factor.
Clin Dev Immunol. 2012:9505032012. View Article : Google Scholar
|
|
23
|
Brightbill HD, Libraty DH, Krutzik SR, et
al: Host defense mechanisms triggered by microbial lipoproteins
through toll-like receptors. Science. 285:732–736. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Straus DS and Glass CK: Anti-inflammatory
actions of PPAR ligands: new insights on cellular and molecular
mechanisms. Trends Immunol. 28:551–558. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Moraes LA, Piqueras L and Bishop-Bailey D:
Peroxisome proliferator-activated receptors and inflammation.
Pharmacol Ther. 110:371–385. 2006. View Article : Google Scholar
|
|
26
|
Mei CL, He P, Cheng B, Liu W, Wang YF and
Wan JJ: Chlamydia pneumoniae induces macrophage-derived foam cell
formation via PPAR alpha and PPAR gamma-dependent pathways. Cell
Biol Int. 33:301–308. 2009. View Article : Google Scholar
|
|
27
|
Liu YL, Lu J, Shi J, et al: Increased
expression of the peroxisome proliferator-activated receptor gamma
in the immune system of weaned pigs after Escherichia coli
lipopolysaccharide injection. Vet Immunol Immunopathol. 124:82–92.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pascual G, Fong AL, Ogawa S, et al: A
SUMOylation-dependent pathway mediates transrepression of
inflammatory response genes by PPAR-gamma. Nature. 437:759–763.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
de Assis EF, Silva AR, Caiado LFC, et al:
Synergism between platelet-activating factor-like phospholipids and
peroxisome proliferator-activated receptor gamma agonists generated
during low density lipoprotein oxidation that induces lipid body
formation in leukocytes. J Immunol. 171:2090–2098. 2003. View Article : Google Scholar : PubMed/NCBI
|