Introduction
Paclitaxel (Taxol®) is an anti-mitotic
agent that promotes apoptosis in cancer cells through multiple
mechanisms (1); the stabilization
of microtubules, mitotic arrest, and eventually cell death
(2). The ability of Taxol to
inhibit tumor cells has made it an effective chemotherapeutic agent
against numerous cancer types, including ovary, breast, oral, colon
and lung, as well as malignant melanoma (3–8).
Despite impressive initial clinical responses, the
majority of patients eventually develop resistance to Taxol
(9). One mechanism known to be
involved in cancer cell resistance to Taxol and other
microtubule-stabilizing agents, is the high expression of membrane
P-glycoprotein which functions as a drug-efflux pump (10). Other cellular mechanisms include
alterations of tubulin structure (11–13),
changes in the drug-binding affinity of the microtubules (14) and cell cycle deregulation (15,16).
The mechanisms of cancer cell intrinsic and acquired paclitaxel
resistance are currently not well understood.
Cancer cells, unlike normal cells, use glycolysis
with reduced mitochondrial oxidative phosphorylation for energy
production (17). Thus, the
metabolic switch by cancer cells to undergo anaerobic, rather than
aerobic, respiration is preferential for the progression of cancer.
In anaerobic respiration the pyruvate generated from glycolysis is
converted into lactic acid, whereas in aerobic respiration pyruvate
is converted into acetyl-CoA by the action of pyruvate
dehydrogenase (PDH). Pyruvate dehydrogenase kinase 1 (PDK1) can
inactivate PDH by phosphorylation. Inhibition of PDH prevents the
conversion of pyruvate to acetyl-CoA, thus resulting in the switch
of metabolism to anaerobic glycolysis, which has proved to be
advantageous for tumor growth (18).
Dichloroacetate (DCA), an inhibitor of PDK1, has
previously been demonstrated to promote cancer cell apoptosis. DCA
is therefore considered to be a promising antineoplastic agent
(19). By blocking PDK1, DCA
decreases lactate production, and promotes the metabolism of
pyruvate, generated from glycolysis, towards oxidation in the
mitochondria (20). Clinical
trials are underway for the use of DCA in the treatment of
disorders of lactic acid accumulation, as well as numerous types of
cancer (21). It has been reported
that DCA can restore mitochondrial function, thus restoring
apoptosis in cancer cells in vitro, and shrink tumor size in
rats (21). A previous paper
demonstrated that PDK1, as well as other enzymes important for
glucose metabolism, were upregulated in myeloma, and that a
combination of DCA and bortezomib showed cytotoxic effects,
indicating that PDK1 inhibition may serve as a novel therapeutic
approach (22).
In the present study, an essential function of PDK1
in Taxol-induced oral cancer cell apoptosis was reported. Cancer
cells displayed resistance to Taxol treatment following a short
incubation period under hypoxic conditions. In addition, Taxol
treatment upregulated both mRNA and protein expression levels of
PDK1. Taxol-resistant oral cancer cells showed higher expression of
PDK1 as compared with Taxol-sensitive cancer cells. The combination
of the PDK1 inhibitor DCA with Taxol resulted in synergistic
inhibitory effects on Taxol-resistant cancer cells under hypoxic
conditions. This study proposed novel aspects for overcoming Taxol
resistance in oral cancer cells, and will contribute to the
development of clinical therapeutics for patients with cancer.
Materials and methods
Cells and cell culture conditions
OECM-1 and SAS human oral squamous cell carcinoma
(OSCC) cell lines, were purchased from ATCC (Rockefeller, MD, USA).
Briefly, OECM-1 and SAS cells were routinely cultured in Dulbecco’s
modified Eagle’s medium (DMEM; Gibco-BRL, Paisley, UK) containing
10% fetal bovine serum (FBS; HyClone, VT, USA), at 37°C in a humid
atmosphere with 5% CO2. Hypoxic conditions were induced
by incubation of the cells in a sealed Bug-Box anaerobic
workstation (Ruskinn Technology, Ltd., Bridgend, UK). For the
hypoxia experiments, the oxygen levels were maintained at 2%, with
the residual gas mixture being 94.0 to 94.9% N2 and 5%
CO2, as compared with the normoxia experiments, in which
the oxygen levels were maintained at 21%.
Antibodies and reagents
A rabbit monoclonal antibody against PDK1 was used
at a 1:1,000 dilution(#3820; Cell Signaling Technology, Danvers,
MD, USA); a β-Actin antibody was used as a loading control, at a
1:2,000 dilution (sc-1616; Santa Cruz Biotechnology, Santa Cruz,
CA, USA). DCA and Taxol were purchased from Sigma-Aldrich (St.
Louis, MO, USA).
cDNA synthesis and quantitative
polymerase chain reaction (qPCR)
RNA was extracted from oral cancer cells using
TRIzol™ reagent (Invitrogen Life Technologies, Carlsbad, CA, USA).
The cDNA synthesis was performed using a SuperScript First-Standard
Synthesis System for RT-PCR (Invitrogen Life Technologies),
according to the manufacturer’s instructions. qPCR analyses were
performed using Assay-on-Demand primers and the TaqMan Universal
PCR Master Mix reagent (Applied Biosystems, Foster City, NJ, USA).
The samples were analyzed using an ABI Prism® 7700
Sequence Detection System (Applied Biosystems). The primers for the
qPCR were: PDK1 forward, 5′-GCTTATCAGAAACTCCAAAGACTGC-3′ and
reverse, 5′-GCCGGAAAGGTGGGACA-3′. The expression levels of β-actin
were used to normalize the relative expression levels. All
experiments were performed in triplicate.
Western blotting
Cells were harvested and lysed in a buffer
containing 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 2 mM EDTA, 1%
Triton, 1 mM phenylmethanesulfonyl fluoride and Protease Inhibitor
Cocktail (Sigma-Aldrich) for 20 min on ice. Lysates were
centrifuged at 16,000 × 6 at 4°C for 10 min. The supernatants were
collected and the protein concentration was determined by Bradford
assay (Bio-Rad, Hercules, CA, USA). The proteins were then
separated by SDS-PAGE and transferred to a nitrocellulose membrane
(Bio-Rad). Following blocking of the membranes with
phosphate-buffered saline (PBS) with 5% non-fat dry milk for 1 h,
the membranes were incubated with the primary antibodies, in PBS
with 5% non-fat dry milk, overnight at 4–8°C. The membranes were
extensively washed with PBS and incubated with horseradish
peroxidase conjugated secondary anti-mouse antibody or anti-rabbit
antibody (1:2,000, Bio-Rad). Following additional washes with PBS,
the antigen-antibody complexes were visualized using an Enhanced
Chemiluminescence kit (Pierce Biotechnology, Inc., Rockford, IL,
USA).
Cell viability assay
Oral cancer cells were seeded in a 6-well plate, at
a density of 10,000 cells/well in 2 ml DMEM containing 10% FBS.
Following overnight incubation under the same culturing conditions,
each well was refreshed with 2 ml serum-free medium (SFM) for
another day. Following this, the cells were then treated with 2 ml
SFM containing various concentrations of Taxol or DCA. The
drug-containing SFM was refreshed after 2 days, and incubated under
the same conditions for a further 2 days. The cell viability was
assessed using MTT reagent (Sigma-Aldrich), and by measuring the
absorbance at 570 nm using a plate reader. The relative viability
was obtained from the absorbance at 570 nm of both the drug-treated
and untreated OECM-1 cells. The experiment was repeated three
times.
Generation of a Taxol-resistant cell
line
OECM-1 Taxol-resistant cells were obtained by
gradually increasing the concentration of Taxol in the cell culture
medium, followed by selection of Taxol-resistant cells. After
successive Taxol treatments for a duration of 3 months, several
resistant cell clones were generated.
Statistical analysis
The data was analyzed using the unpaired Student’s
t-test. All the data are expressed as the means ± standard error. A
P<0.05 was considered to indicate a statistically significant
difference.
Results
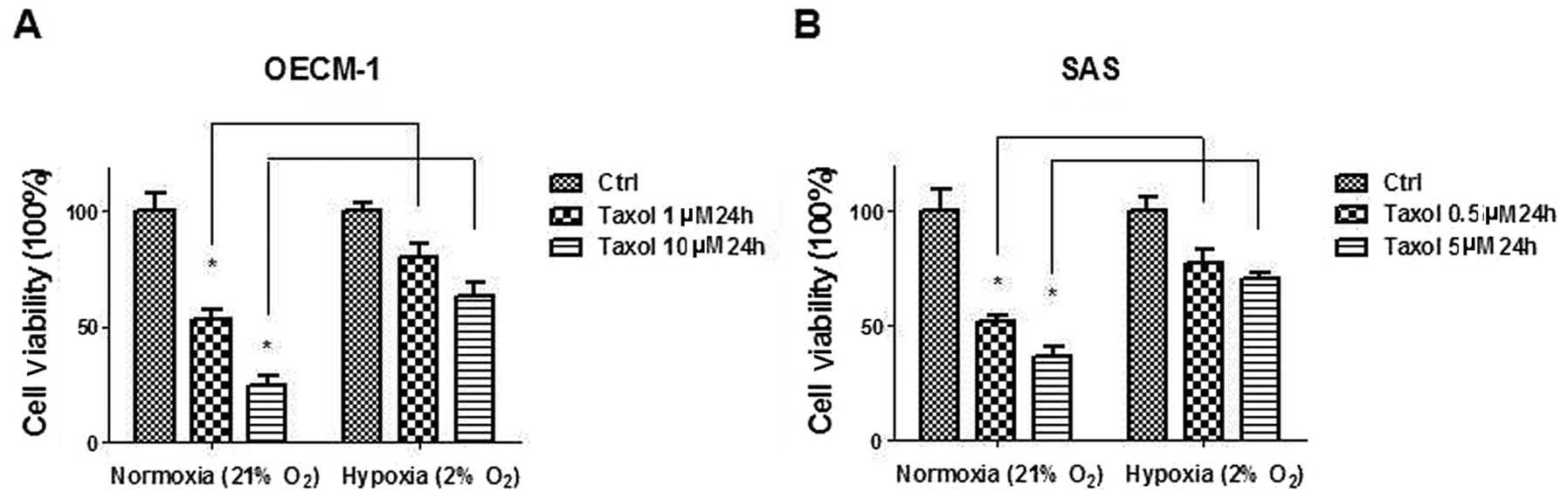
Oral cancer cells acquire resistance to
Taxol under hypoxia
Since the switch from mitochondrial oxidation to
anaerobic glycolysis renders cancer cells resistant to chemotherapy
(23), OECM-1 and SAS human oral
cancer cell lines were cultured under hypoxic conditions for 24 h
followed by measurements of Taxol sensitivity. Under hypoxic
conditions, both cell lines acquired resistance to Taxol at
multiple concentrations, as compared with cells cultured in
normoxic conditions (Fig. 1A and
B). It has previously been reported that low oxygen stabilizes
hypoxia inducible factor-1 α (HIF-1α) which promotes the
transcriptional expression of enzymes that are required for
glycolysis (24). The results of
the present study indicated that HIF-1α-mediated upregulation of
glycolysis may contribute to Taxol resistance in oral cancer
cells.
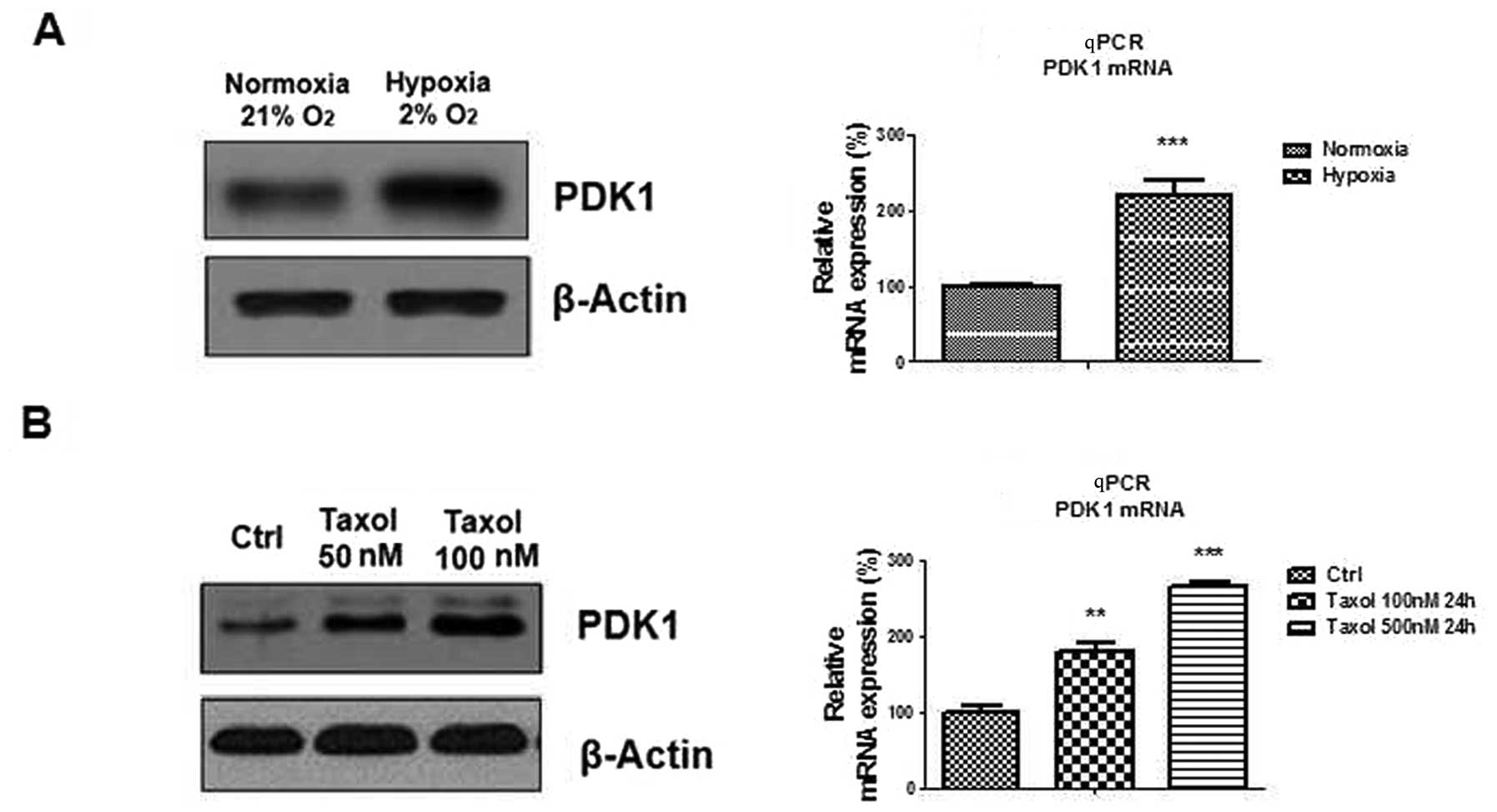
Hypoxia induces PDK1 expression in oral
cancer cells
The protein and mRNA expression levels of pyruvate
dehydrogenase kinase 1 were measured. Both PDK1 mRNA and protein
expression was upregulated by hypoxia (Fig. 2A), suggesting that PDK1 may be
involved in Taxol sensitivity in oral cancer cells.
PDK1 is induced by Taxol in oral cancer
cells
It has been reported that treatment with Taxol can
upregulate and increase the activity of lactate dehydrogenase A
(LDHA) expression, which contributes towards Taxol resistance in
breast cancer cells (25). Since
both LDHA and PDK1 are key enzymes of glycolysis, it was
hypothesized that PDK1 may participate in Taxol resistance. PDK1
protein and mRNA expression was measured in oral cancer cells in
response to Taxol treatments. PDK1 was significantly upregulated by
Taxol treatment at multiple concentrations (Fig. 2B), indicating that PDK1 may be
induced by Taxol treatment and hypoxia. These results may provide a
potential mechanism for Taxol resistance.
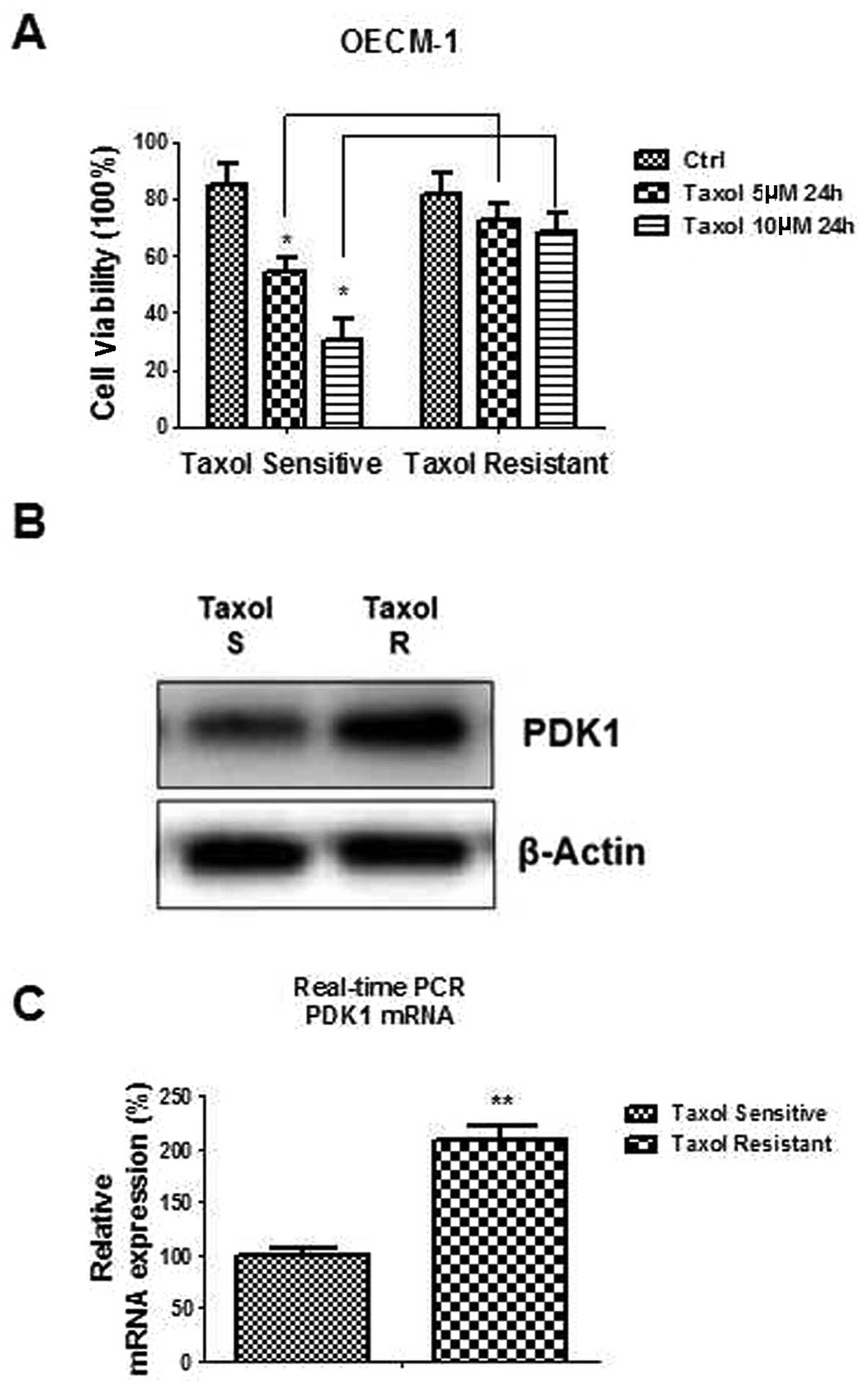
Taxol resistant cells exhibit upregulated
PDK1
OECM-1 Taxol-resistant cells were generated by
gradually increasing concentrations of Taxol in the cell culture
medium, followed by selection of Taxol-resistant cells. Several
resistant cell clones were developed after successive Taxol
treatments for 3 months. OECM-1 Taxol-resistant cells had ~70%
viability under 5 and 10 μM Taxol treatments, which was much higher
as compared with Taxol-sensitive cells (Fig. 3A). The mRNA and protein expression
of PDK1 was measured in both Taxol-sensitive and Taxol-resistant
cells. Fig. 3B and C showed that
both the PDK1 mRNA and protein expression levels were increased in
Taxol-resistant cells, as compared with Taxol-sensitive cells.
These results suggest that PDK1 is important in cancer cell Taxol
resistance.
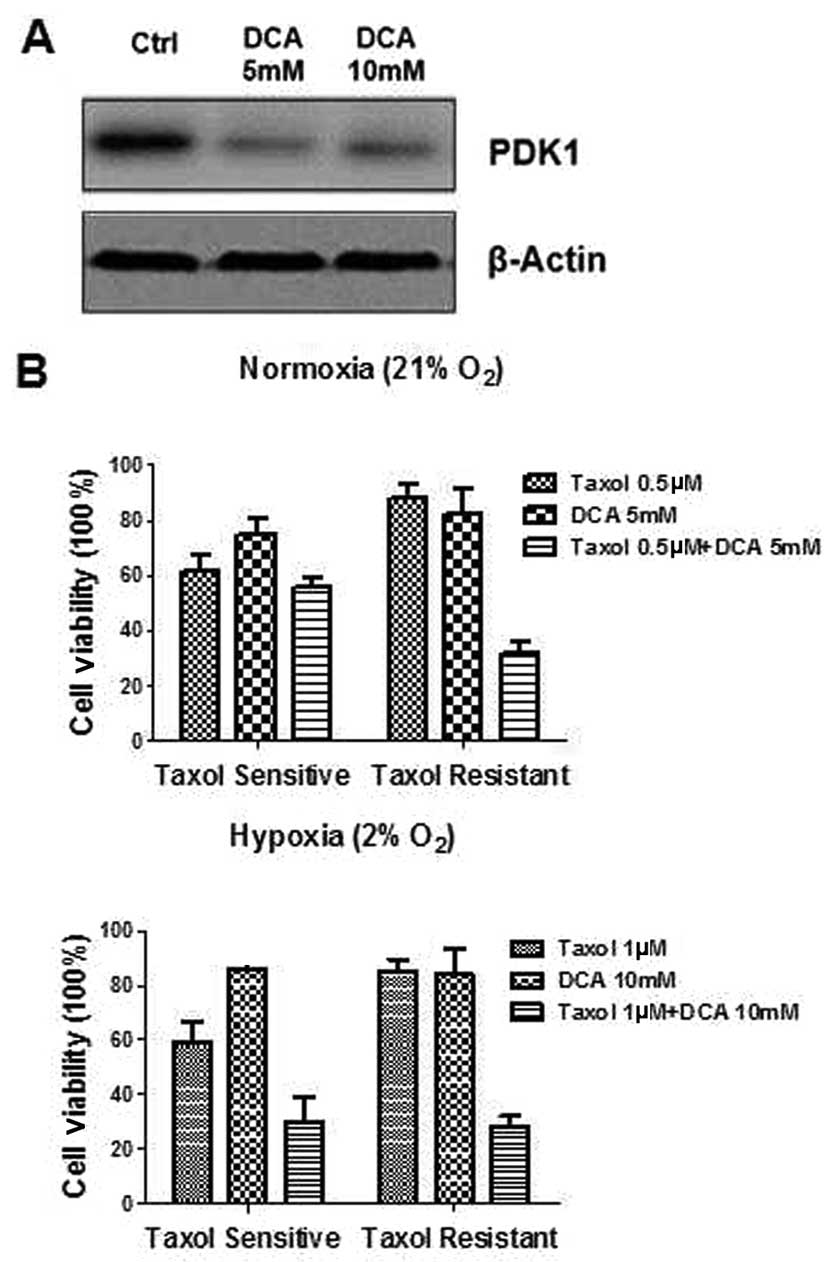
The combination of DCA and Taxol has
synergistic effects on Taxol-resistant oral cancer cells
DCA, an inhibitor of PDK, has been demonstrated to
inhibit glycolysis and promote cancer cell apoptosis. OECM-1 cells
were treated with 5 and 10 mM concentrations of DCA for 24 h, after
which, the PDK1 protein and mRNA expression levels were measured.
Figure 4A shows protein expression
levels of PDK1 were significantly suppressed by DCA at 5 and 10 mM,
as compared with the controls. To further explore the possible
biological significance of DCA in Taxol resistance, oral cancer
cells were treated with Taxol alone, DCA alone, or a combination of
Taxol and DCA under hypoxia. The combination of Taxol and DCA
demonstrated a synergistic inhibitory effect on both
Taxol-sensitive and resistant cells under hypoxia (Fig. 4B). However, a combination of Taxol
and DCA did not exhibit a synergistically inhibitory effect in
Taxol-sensitive cells under normoxia. (Fig. 4B). A possible explanation for this
may be because the PDK1 expression in Taxol-sensitive cells was not
altered when the cells were conditioned under the normal oxygen
conditions, resulting in a non-synergistic effect.
Discussion
Taxol is a widely used chemotherapeutic agent for
the treatment of several types of cancers, including breast cancer
(26). Taxol resistance may result
in the recurrence and metastasis of cancer, ultimately increasing
the risk of mortality (27).
Although extensive research has been carried out in regards to the
resistance of cancer cells to Taxol, the specific mechanisms
involved are still poorly understood (28). The ability to reduce Taxol
resistance would be of benefit to cancer patients, demonstrating
the importance of research into the underlying mechanisms of how
chemoresistance may arise. Numerous studies support the concept
that dysregulated cellular metabolism is linked to drug resistance
in cancer therapy (23,29). It has been previously reported that
Taxol-resistant breast cancer cells have an upregulation of LDHA
expression and activity, as compared with Taxol-sensitive cells
(25), indicating hyperactive
glucose metabolism in Taxol-resistant cells.
The Warburg effect refers to the difference in
metabolism between normal and cancerous tissues; tumors consume
more glucose and produce lactate, even in the presence of adequate
oxygen (17). It has been
understood that energy metabolism in cancer cells depends on
glycolysis, followed by lactate production, rather than oxidative
phosphorylation in mitochondria (30). It has been previously reported that
several oncogenes, including AKT, MYC and ErbB2, as well as HIF-1
are able to regulate aerobic glycolysis, which contributes to
chemoresistance (24,29,31,32).
Myc induces the expression of genes regulating glycolysis, such as
GLUT1, HK2, Enolase 1 and LDHA (32). Under hypoxic conditions, HIF-1 also
induces the expression of glycolytic enzymes, such as GLUT1,
Enolase 1, LDHA and PDK1 (24).
PDK1 is a serine/threonine kinase which phosphorylates and inhibits
mitochondrial PDH, the enzyme responsible for the conversion of
pyruvate to acetyl-CoA. PDK1 is a key regulator of the Warburg
effect (33). DCA, a PDK inhibitor
has been proposed as an anticancer agent which targets glycolysis.
As well as the regulation of glycolysis, DCA has been shown to
possess other anti-cancer activities, including induction of cell
cycle arrest, and depolarization of the hyperpolarized inner
mitochondrial membrane (34). Oral
administration of DCA was previously reported to show good
bioavailability and encouraging phase I/II clinical trials for its
use in patients with brain and non-small cell lung cancer (35). A new selective PDK1 inhibitor,
AZD7545, is also expected to undergo a clinical trial (36).
In the present study, PDK1 was reported as a novel,
putative target for overcoming Taxol resistance in oral cancer
cells cultured under hypoxic conditions. Both low oxygen and
treatment of cells with Taxol-induced PDK1 expression, rendering
cancer cells more vulnerable to the PDK1 inhibitor, DCA. When
Taxol-resistant cells were pre-incubated in a low oxygen
environment, PDK1 expression was induced, resulting in a
synergistic therapeutic effect with Taxol. The present study was
focused on PDK1, which is one of the key metabolic enzymes in
glycolysis. It remains to be determined whether other enzymes, such
as GLUT1, HK2 and LDHA, are involved in Taxol resistance in oral
cancer cells. More importantly, the mechanism by which Taxol
induces PDK1 expression is still under investigation. Our future
studies will focus on the selection of other PDK1 inhibitors, and
will explore the mechanisms by which Taxol induces PDK1 expression
in human oral cancer cells. In conclusion, the results of the
present study highlight the importance of PDK1 in its role in Taxol
resistance, and may allow for possible therapeutic interventions in
patients that have developed a resistance to Taxol.
Acknowledgements
This work was financially supported by the Key
Technology Research and Development Program of Heilongjiang
Province (no. GC12C305-3).
References
|
1
|
Orr GA, Verdier-Pinard P, McDaid H and
Horwitz SB: Mechanisms of Taxol resistance related to microtubules.
Oncogene. 22:7280–7295. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Marcus AI, Peters U, Thomas SL, et al:
Mitotic kinesin inhibitors induce mitotic arrest and cell death in
Taxol-resistant and -sensitive cancer cells. J Biol Chem.
280:11569–11577. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yang YI, Lee KT, Park HJ, et al:
Tectorigenin sensitizes paclitaxel-resistant human ovarian cancer
cells through downregulation of the Akt and NFκB pathway.
Carcinogenesis. 33:2488–2498. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lai D, Ho KC, Hao Y and Yang X: Taxol
resistance in breast cancer cells is mediated by the hippo pathway
component TAZ and its downstream transcriptional targets Cyr61 and
CTGF. Cancer Res. 71:2728–2738. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ledwitch K, Ogburn R, Cox J, et al: Taxol:
efficacy against oral squamous cell carcinoma. Mini Rev Med Chem.
13:509–521. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xu R, Sato N, Yanai K, et al: Enhancement
of paclitaxel-induced apoptosis by inhibition of mitogen-activated
protein kinase pathway in colon cancer cells. Anticancer Res.
29:261–270. 2009.PubMed/NCBI
|
|
7
|
Groen HJ, Fokkema E, Biesma B, et al:
Paclitaxel and carboplatin in the treatment of small-cell lung
cancer patients resistant to cyclophosphamide, doxorubicin, and
etoposide: a non-cross-resistant schedule. J Clin Oncol.
17:927–932. 1999.PubMed/NCBI
|
|
8
|
González Cao M, Viteri S, Garrán C, et al:
Response of resistant melanoma to a combination of weekly
paclitaxel and bevacizumab. Clin Transl Oncol. 9:119–120. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yusuf RZ, Duan Z, Lamendola DE, Penson RT
and Seiden MV: Paclitaxel resistance: molecular mechanisms and
pharmacologic manipulation. Curr Cancer Drug Targets. 3:1–19. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gottesman MM and Pastan I: Biochemistry of
multidrug resistance mediated by the multidrug transporter. Annu
Rev Biochem. 62:385–427. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kavallaris M, Kuo DY, Burkhart CA, et al:
Taxol-resistant epithelial ovarian tumors are associated with
altered expression of specific beta-tubulin isotypes. J Clin
Invest. 100:1282–1293. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Panda D, Miller HP, Banerjee A, Ludueña RF
and Wilson L: Microtubule dynamics in vitro are regulated by the
tubulin isotype composition. Proc Natl Acad Sci USA.
91:11358–11362. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Martello LA, Verdier-Pinard P, Shen HJ, et
al: Elevated levels of microtubule destabilizing factors in a
Taxol-resistant/dependent A549 cell line with an alpha-tubulin
mutation. Cancer Res. 63:1207–1213. 2003.PubMed/NCBI
|
|
14
|
Gonçalves A, Braguer D, Kamath K, et al:
Resistance to Taxol in lung cancer cells associated with increased
microtubule dynamics. Proc Natl Acad Sci USA. 98:11737–11742. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tan M, Jing T, Lan KH, et al:
Phosphorylation on tyrosine-15 of p34(Cdc2) by ErbB2 inhibits
p34(Cdc2) activation and is involved in resistance to taxol-induced
apoptosis. Mol Cell. 9:993–1004. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lu J, Tan M, Huang WC, et al: Mitotic
deregulation by survivin in ErbB2-overexpressing breast cancer
cells contributes to Taxol resistance. Clin Cancer Res.
15:1326–1334. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Warburg O: On respiratory impairment in
cancer cells. Science. 124:269–270. 1956.PubMed/NCBI
|
|
18
|
Jang M, Kim SS and Lee J: Cancer cell
metabolism: implications for therapeutic targets. Exp Mol Med.
45:e452013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Saed GM, Fletcher NM, Jiang ZL, et al:
Dichloroacetate induces apoptosis of epithelial ovarian cancer
cells through a mechanism involving modulation of oxidative stress.
Reprod Sci. 18:1253–1261. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sutendra G and Michelakis ED: Pyruvate
dehydrogenase kinase as a novel therapeutic target in oncology.
Front Oncol. 3:382013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Michelakis ED, Webster L and Mackey JR:
Dichloroacetate (DCA) as a potential metabolic-targeting therapy
for cancer. Br J Cancer. 99:989–994. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fujiwara S, Kawano Y, Yuki H, et al: PDK1
inhibition is a novel therapeutic target in multiple myeloma. Br J
Cancer. 108:170–178. 2013.PubMed/NCBI
|
|
23
|
Zhao Y, Butler EB and Tan M: Targeting
cellular metabolism to improve cancer therapeutics. Cell Death Dis.
4:e5322013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fulda S and Debatin KM: HIF-1-regulated
glucose metabolism: a key to apoptosis resistance? Cell Cycle.
6:790–792. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhou M, Zhao Y, Ding Y, et al: Warburg
effect in chemosensitivity: targeting lactate dehydrogenase-A
re-sensitizes taxol-resistant cancer cells to taxol. Mol Cancer.
9:332010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bryan M, Pulte ED, Toomey KC, et al: A
pilot phase II trial of all-trans retinoic acid (Vesanoid) and
paclitaxel (Taxol) in patients with recurrent or metastatic breast
cancer. Invest New Drugs. 29:1482–1487. 2011. View Article : Google Scholar
|
|
27
|
Ishibashi M, Nakayama K, Yeasmin S, et al:
A BTB/POZ gene, NAC-1, a tumor recurrence-associated gene, as a
potential target for Taxol resistance in ovarian cancer. Clin
Cancer Res. 14:3149–3155. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang X, Pan L, Mao N, et al: Cell-cycle
synchronization reverses Taxol resistance of human ovarian cancer
cell lines. Cancer Cell Int. 13:772013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ding Y, Liu Z, Desai S, et al: Receptor
tyrosine kinase ErbB2 translocates into mitochondria and regulates
cellular metabolism. Nat Commun. 3:12712012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zheng J: Energy metabolism of cancer:
Glycolysis versus oxidative phosphorylation (Review). Oncol Lett.
4:1151–1157. 2012.PubMed/NCBI
|
|
31
|
Elstrom RL, Bauer DE, Buzzai M, et al: Akt
stimulates aerobic glycolysis in cancer cells. Cancer Res.
64:3892–3899. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fan Y, Dickman KG and Zong WX: Akt and
c-Myc differentially activate cellular metabolic programs and prime
cells to bioenergetic inhibition. J Biol Chem. 285:7324–7333. 2010.
View Article : Google Scholar :
|
|
33
|
Kim JW, Tchernyshyov I, Semenza GL and
Dang CV: HIF-1-mediated expression of pyruvate dehydrogenase
kinase: a metabolic switch required for cellular adaptation to
hypoxia. Cell Metab. 3:177–185. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Madhok BM, Yeluri S, Perry SL, Hughes TA
and Jayne DG: Dichloroacetate induces apoptosis and cell-cycle
arrest in colorectal cancer cells. Br J Cancer. 102:1746–1752.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Michelakis ED, Sutendra G, Dromparis P, et
al: Metabolic modulation of glioblastoma with dichloroacetate. Sci
Transl Med. 2:31ra342010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kato M, Li J, Chuang JL and Chuang DT:
Distinct structural mechanisms for inhibition of pyruvate
dehydrogenase kinase isoforms by AZD7545, dichloroacetate, and
radicicol. Structure. 15:992–1004. 2007. View Article : Google Scholar : PubMed/NCBI
|