Introduction
Gastric cancer is a common malignancy on mainland
China, with high rates of incidence and mortality (1). The diagnosis of novel gastric cancer
cases in China comprise 40% of cases diagnosed worldwide every year
(2,3). In addition, >80% of patients with
gastric cancer in developing countries, including China, are
diagnosed at advanced stages of the disease, resulting in poor
prognoses (4,5). Despite major advances in gastric
cancer research over the past few decades, an effective gastric
cancer therapy has remained to be developed (6). However, a common feature of gastric
cancer is an uncontrolled cell growth and proliferation mechanism.
The identification of novel molecules associated with gastric
cancer cell proliferation may aid the development of more effective
therapies. The present study therefore aimed to identify additional
molecules crucially involved in gastric cancer cell growth.
A diverse array of G protein-coupled receptors
(GPRs; also known as GPCRs) with numerous functions have been
implicated in the pathophysiology of multiple types of cancer and
are under investigation as potential drug targets (7–9).
GPRs are critical mediators of signal transduction and are involved
in modulating the cellular proliferation of diverse types of
cancer. Certain GPRs, including lysophosphatidic acid,
acetylcholine, prostaglandin E2 and bradykinin receptors, induce
the growth and proliferation of head and neck squamous-cell
carcinomas (10,11). Proteolytic activation of
protease-activated receptors (PARs, a GPR subtype) by thrombin
stimulates the proliferation of breast carcinoma cells via the
activation of epidermal growth factor signaling (12). Breast and ovarian cancer cell
proliferation was also reported to be associated with endothelin A
subtype receptor (ETAR, a GPR) (13), highlighting the prominent role of
GPRs in cancer cell proliferation and growth.
G protein-coupled receptor 137 (GPR137), also known
as transmembrane 7 superfamily member 1-like 1 protein (TM7SF1L1),
C11orf4 or GPR-137A, and is a 417 amino acid member of the GPR-137
family of membrane proteins. The GPR137 family comprises three
alternatively spliced isoforms: GPR137A (GPR137), GPR137B and
GPR137C. The GPR137B isoform has been studied extensively and is
upregulated in the kidney during development (14). GPR137B was also found to be
abundant in the liver and heart but little or no expression was
detected in the intestine, spleen or other tissues in the rat
(15). Unlike GPR137B, GPR137 and
GPR137C have rarely been studied. GPR137 was identified in 2003 by
searching the GenBank genomic databases (16). Northern blot analysis revealed that
GPR137 was detected in the hippocampus, but not in the
hypothalamus, midbrain, thalamus, pons or basal forebrain. The role
of GPR137 in human cancers is poorly understood, and the function
of GPR137 and its regulatory mechanism in human cancer has remained
to be elucidated.
Gene knockdown using small interfering RNA (siRNA)
represents a useful tool for the assessment of the functional
importance of cancer-associated genes in vitro. The present
study therefore aimed to investigate the role of GPR137 in gastric
cancer cell growth using RNA interference (RNAi).
Materials and methods
Cell lines and cell culture
The gastric cancer cell lines MKN28, SGC-7901,
MGC80-3 and AGS were obtained from the American Type Culture
Collection (Manassas, VA, USA). Cells were maintained in RPMI-1640
supplemented with 10% heat-inactivated fetal bovine serum (FBS) and
penicillin/streptomycin (All from Gibco-BRL, Invitrogen Life
Technologies, Carlsbad, CA, USA) at 37°C in a humidified atmosphere
of 5% CO2.
RNA extraction and reverse transcription
quantitative polymerase chain reaction (RT-qPCR)
Total RNAs of cultured cells were extracted using
TRIzol solution (Invitrogen Life Technologies, Carlsbad, CA, USA)
24 h following treatment. The cDNA was immediately reverse
transcribed from isolated RNA using the SuperScript III
First-Strand Synthesis System (Invitrogen Life Technologies).
GPR137 mRNA expression was evaluated by qPCR analysis using the
SYBR Premix Ex Taq™ Perfect Real-Time (Takara Bio, Inc., Otsu,
Japan) on an ABI Prism 7500 real-time system (Applied Biosystems,
Life Technologies, Foster City, CA, USA). GAPDH was used as the
input reference. The primers used were as follows: GPR137 forward,
5′-ACCTGGGGAACAAAGGCTAC-3′ and reverse, 5′-TAGGACCGAGAGGCAAAGAC-3′;
GAPDH forward, 5′-GTGGACATCCGCAAAGAC-3′ and reverse,
5′-AAAGGGTGTAACGCAACTA-3′.
Relative mRNA was determined using the formula
2−ΔΔCT (with CT being the cycle threshold), where ΔCT =
[CT (target gene) - CT (GAPDH)] as described previously (17).
Construction of GPR137 short hairpin RNA
(shRNA)-expressing lentivirus
To construct cell lines stably expressing GPR137
shRNA, an shRNA (5′-GAACAAAGGCTA
CCTGGTATTCTCGAGAATACCAGGTAGCCTTTGTTCT TTTTT-3′) was inserted into
the pFH-L plasmid (Shanghai Hollybio, Shanghai, China). A
non-silencing siRNA (5′-TTCTCCGAACGTGTCACGT-3′) was used as a
control. The lentivirus-based shRNA-expressing vectors were
constructed, confirmed by DNA sequencing and named pFH-L-shGPR137
and pFH-L-shCon. For the transfection assay, AGS and MGC80-3 cells
(5×104 cells/well) were seeded into six-well plates and
cultured for 72 h to reach 90% confluence. At 2 h prior to
transfection, the culture medium was replaced with FBS-free
RPMI-1640. The plasmid mixture containing pFH-L-shGPR137 (or
pFH-L-shCon) and the pVSVG-I/pCMVΔR8.92 packaging vectors (Shanghai
Hollybio), as well as Lipofectamine® 2000 (Invitrogen
Life Technologies) was added to the cells. Five hours following
incubation, the culture medium was replaced with RPMI-1640
containing 10% FBS. The lentiviral constructs (Lv-shGPR137 or
Lv-shCon) were harvested 48 h following transfection and purified
by ultra-centrifugation as previously described (18,19).
The lentivirus contained green fluorescent protein (GFP), and
therefore the viral titer was determined by counting the number of
GFP-expressing cells under an Olympus BX50 Brightfield/Fluorescence
microscope (Olympus Corp., Tokyo, Japan) 96 h following infection,
as previously described (20).
Western blot analysis
Samples were homogenized in western blot analysis
buffer containing 10 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% (v/v)
Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 5 mM EDTA, 1 mM
phenylmethanesulfonylfluoride, 0.28 kU/l aprotinin, 50 mg/l
leupeptin, 1 mM benzamidine, and 7 mg/l pepstain A (all purchased
from Sigma-Aldrich, St. Louis, MO, USA). The homogenate was
subsequently centrifuged at 12,000 × g for 15 min at 4°C and the
supernatant was retained and preserved at −80°C for later use. The
protein concentration was determined using a bicinchoninic acid
assay kit (Pierce Biotechnology, Inc., Thermo Fisher Scientific,
Rockford, IL, USA) and each sample was subjected to 10% SDS-PAGE.
Proteins were transferred onto nitrocellulose membranes (EMD
Millipore, Billerica, MA, USA) on a semi-dry electrotransferring
unit (Bio-Rad Laboratories, Hercules, CA, USA), and incubated with
rabbit anti-GPR137 polyclonal antibodies (11929-1-AP; 1:500;
Proteintech Group Inc., Chicago, IL, USA) in Tris-buffered saline
containing 0.1% Tween-20 (TBST; Amresco, Solon, OH, USA) and 5%
non-fat dry milk overnight at 4°C. Following overnight incubation
with the primary antibodies, membranes were washed with TBST and
incubated with horseradish peroxidase-conjugated goat anti-rabbit
and goat anti-mouse antibodies (sc-2054 and sc-2005; 1:5,000; Santa
Cruz Biotechnology, Inc., Dallas, TX, USA) in TBST for 3 h.
Immunoreactivity was detected using enhanced chemiluminescent
autoradiography (ECL kit; GE Healthcare Life Sciences, Little
Chalfont, UK). The membranes were re-probed with mouse anti-GAPDH
monoclonal antibodies (sc-365062; 1:3,000; Santa Cruz
Biotechnology, Inc.) following stripping.
MTT assay
Cell proliferation was determined using a
colorimetric assay with MTT. The MTT assay measures the conversion
of MTT to insoluble formazan by the dehydrogenase enzymes of the
intact mitochondria of living cells. Following treatment with or
without the appropriate plasmid, AGS and MGC80-3 cells were seeded
in 96-well plates (104 cells/well). Cells were allowed
to attach overnight and cell proliferation was evaluated over five
days by measuring the conversion of MTT to formazan crystals.
Briefly, 10 μl MTT reagent [5 mg/ml in phosphate-buffered saline
(PBS); Sigma-Aldrich] was added to the cells and incubated for 4 h
at 37°C. The medium was removed and 200 μl isopropanol was added.
The amount of formazan crystals formed directly correlated with the
number of viable cells. The reaction product was quantified by
measuring the absorbance at 595 nm using an ELISA plate reader
(Model 2550; Bio-Rad Laboratories, Inc). Experiments were performed
in triplicate.
Plate colony formation assay
Cells (104 cells/well) were seeded into
six-well plates three days following lentivirus infection. The
medium was replaced at two-day intervals. Following six days of
culture at 37°C, cells were washed with PBS and fixed with 4%
paraformaldehyde (Sigma-Aldrich) for 30 min at room temperature.
The fixed cells were then stained with freshly prepared diluted
Giemsa (Merck KGaA, Darmstadt, Germany) for 10 min, washed with
water and air-dried. The total number of colonies with >50 cells
was counted using a Olympus BX50 Brightfield/Fluorescence
microscope and an Olympus CH-2 Binocular Light microscope (Olympus
Corp.).
Fluorescence-activated cell sorting
(FACS) analysis
The DNA contents of each cell cycle phase is
reflected by variations in propidium iodide (PI) fluorescence
intensities. The cell cycle distribution of Lv-shGPR137- or
Lv-shCon-infected cells was analyzed by flow cytometry following PI
staining as previously described (21). In brief, cells were collected 96 h
after lentiviral infection and seeded into six-well plates
(2×105 cells/well). Cells were allowed to attach
overnight prior to collection. Following washing with ice-cold PBS,
cells were suspended in ~0.5 ml 70% cold ethanol and kept at 4°C
for 30 min. The cells were subsequently treated with 100 mg/ml
DNase-free RNase (Sigma-Aldrich) and incubated for 30 min at 37°C
prior to the addition of PI (50 mg/ml; Sigma-Aldrich) directly to
the cell suspension. The suspension was filtered through a 50-mm
nylon mesh, and a total of 10,000 stained cells were subjected to
flow cytometric analysis (FACSCalibur; BD Biosciences, San Jose,
CA, USA).
Statistical analysis
Data were analyzed using GraphPad Prism software
version 6.00 for Windows (GraphPad Software, Inc., San Diego, CA,
USA). Values are expressed as the mean ± standard deviation.
Statistical significance between different groups was determined by
repeated-measure analysis of variance. P<0.05 was considered to
indicate a statistically significant difference between values.
Results
GPR137 is differentially expressed
amongst gastric cancer cell lines
In order to investigate the role of GRP137 in
gastric cancer, the expression levels of GPR137 were assessed in a
subset of gastric cancer cell lines, including MKN28, SGC-7901,
MGC80-3 and AGS. When normalized to the expression levels of GRP137
in MKN28 cells, GPR137 was observed to be differentially expressed
amongst the four cell lines. The mRNA expression of GPR137 was
highest in AGS cells, followed by MGC80-3 cells, and the lowest
GPR137 mRNA expression levels were observed in SGC-7901 cells
(Fig. 1A). As shown in Fig. 1B, western blot analysis revealed
analogous results, indicating that GPR137 protein was expressed at
high levels in MGC80-3 and AGS cells and at lower levels in MKN28
cells. mRNA levels of GPR137 in SGC-7901 cells were relatively low
(Fig. 1A); however, the protein
levels of GPR137 were markedly higher. These results revealed the
differential expression profile of GPR137 amongst gastric cancer
cell lines.
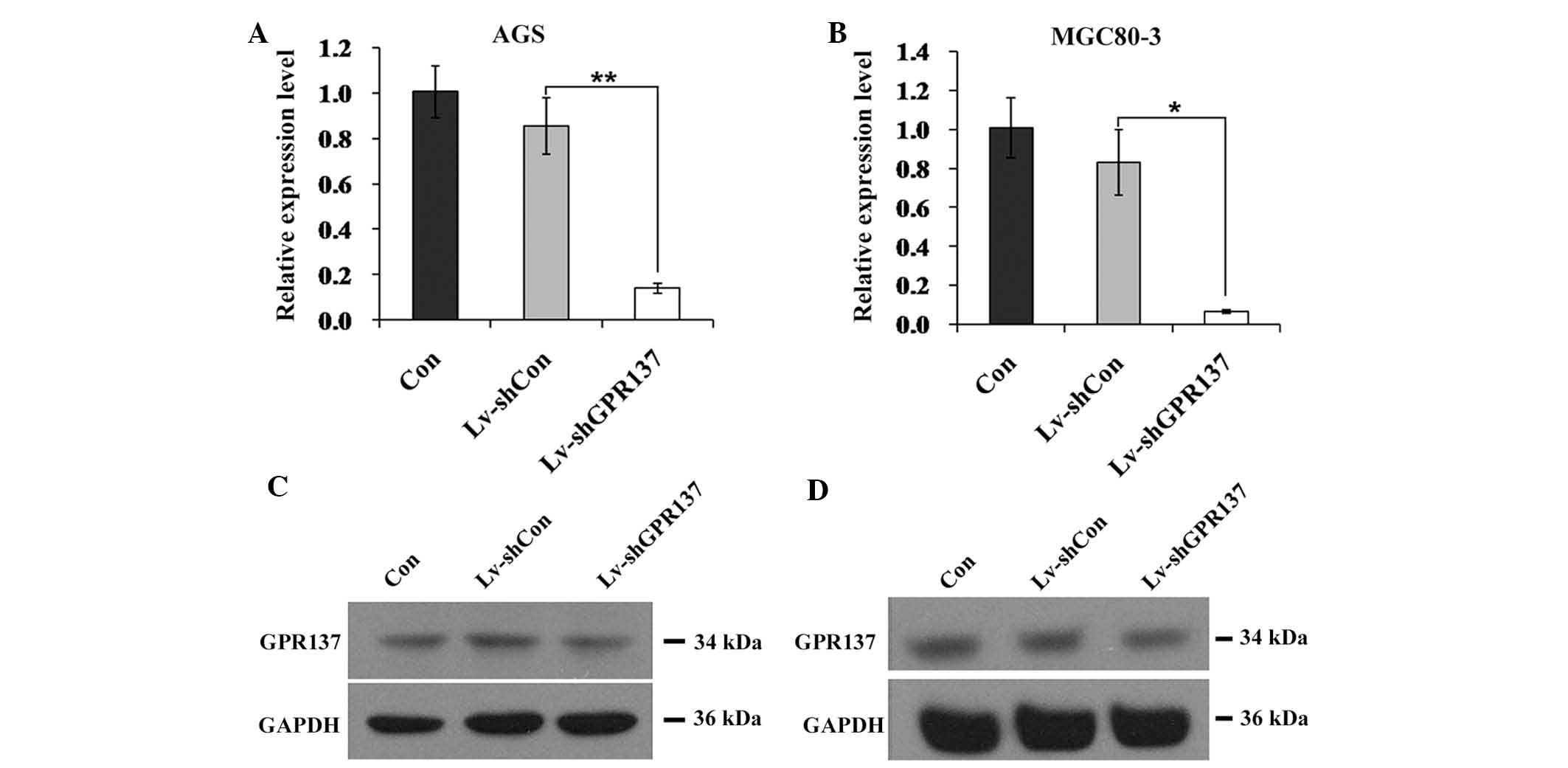
Expression of GPR137 is suppressed by
infection with Lv-shGPR137 in gastric cancer cells
Due to the higher expression levels of GPR137 in
MGC80-3 and AGS cells, and these cell lines were selected for
subsequent analysis. To suppress the expression of GPR137 in
gastric cancer cells, a lentivirus that stably expressed
GPR137-specific siRNA (Lv-shGPR137) was constructed and infected
into the two cell lines (multiplicity of infection, 20). Infection
efficiency was ~90% in the two cell lines (Fig. 2). Furthermore, compared with the
control vector Lv-shCon, the positive infection rate and the GPR137
mRNA expression levels in Lv-shGPR137-infected AGS cells were
significantly decreased (P<0.01; Fig. 3A). Analogous results were obtained
for MGC80-3 cells (P<0.05; Fig.
3B). Concomitantly, the protein expression levels of GPR137
were markedly reduced in AGS and MGC80-3 cells following infection
with Lv-shGPR137 (Fig. 3C and D).
The expression of GPR137 was therefore successfully inhibited by
infection with Lv-shGPR137 in gastric cancer cells.
Knockdown of GPR137 inhibits gastric
cancer cell growth
To investigate the role of GPR137 in gastric cancer
cell growth, cell viability and colony formation assays were
conducted. MGC80-3 and AGS cells were infected with Lv-shGPR137 or
Lv-shCon. In the cell viability assay, cell numbers were evaluated
on days one to five of culture. The number of cells in the control
and Lv-shCon groups was similar, whereas cell proliferation was
markedly reduced in AGS cells infected with Lv-shGPR137
(P<0.001; Fig. 4A). The
proliferation rate of MGC80-3 cells was also decreased following
GPR137 knockdown (P<0.001; Fig.
4B). On day five, the proliferation rate of cells in the
Lv-shGPR137 groups was markedly reduced, by 49.2% in AGS cells and
62.2% in MGC80-3 cells, compared with that of the Lv-shCon groups.
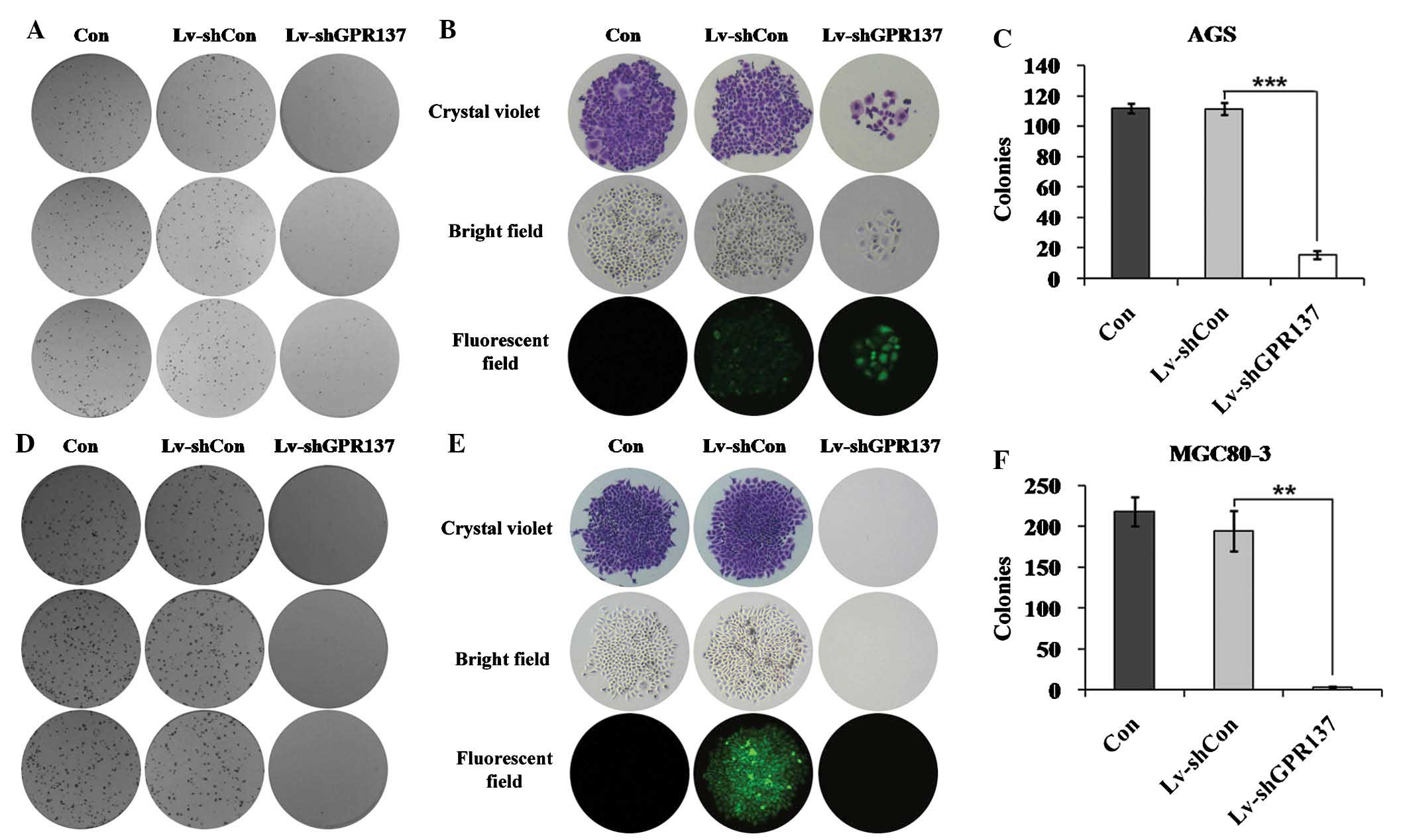
The colony formation assay revealed that AGS cells failed to form
as many colonies as the control and Lv-shCon groups following
infection with Lv-shGPR137 (Fig. 5A
and B). A mean of 15 colonies were generated in
Lv-shGPR137-infected AGS cells, compared with a mean of 112
colonies in the control group and 111 colonies in the Lv-shCon
group (P<0.001; Fig. 5C). The
number of colonies was also reduced in MGC80-3 cells following
GPR137 knockdown (Fig. 5D and E).
Almost no colonies were observed in Lv-shGPR137-infected MGC80-3
cells compared with 218 colonies in the control group and 194
colonies in the Lv-shCon group (P<0.01; Fig. 5F). These results indicated that
knockdown of GPR137 decreased gastric cancer cell
proliferation.
GPR137 depletion arrests cell cycle
progression in gastric cancer cells
To assess the effects of GPR137 depletion on cell
cycle distribution, MGC80-3 cells infected with Lv-shCon or
Lv-shGPR137 were synchronized by serum starvation for 72 h. The
culture medium was subsequently replaced with complete medium
containing 10% FBS and cell cycle progression was assayed 24 h
later by flow cytometry. The number of MGC80-3 cells at each phase
(G0/G1, S or G2/M) was
differentially distributed amongst the three groups (Con, Lv-shCon
and Lv-shGPR137; Fig. 6A). At each
phase, cells were similarly distributed amongst the non-infected
and Lv-shCon-infected MGC80-3 cells (Fig. 6B). However, a greater proportion of
cells were halted in G2/M phase (~22.6%) when GPR137 was depleted
(P<0.05). The proportion of cells in G0/G1
phase was significantly reduced when GPR137 was depleted,
accounting for only 44.41%, compared to 54.31% in the control and
Lv-shCon groups (P<0.05). These results suggested that the cell
cycle was arrested following GPR137 knockdown, which indicated that
GPR137 may have a significant role in gastric cancer cell cycle
progression.
Discussion
The development of gastric carcinogenesis is complex
and multifactorial, involving Helicobacter pylori, as well
as environmental and genetic factors (22). The development of gastric cancer
occurs following the accumulation of multiple genetic and
epigenetic alterations during the lifetime of the individual in
question (22). These changes
eventually trigger extracellular signals to become intracellular
signals. The majority of patients with gastric cancer present with
late-stage disease and have poor prognoses; therefore, the
identification of aberrantly expressed proteins associated with
gastric carcinogenesis was required. GPR137 was previously
identified as a novel G protein-coupled receptor and its transcript
was detected in diverse brain tissues (16). However, the role of GPR137 in
organs outside of the human brain remained to be elucidated. The
results of the present study demonstrated that GPR137 was able to
mediate gastric cancer cell growth in vitro. Depletion of
GPR137 by lentivirus-delivered shRNA markedly reduced the number of
cells detected using a cell viability assay. Concomitantly, gastric
cancer cells failed to maintain colony formation ability following
infection with Lv-shGPR137. In addition, cell cycle phase
distribution was altered.
Cell viability assays were used to assess the cell
proliferation rate which also gave an indication of cell numbers
(23). Similarly, colony formation
assays facilitated the assessment of cell growth capability under
anchorage-independent conditions, and are closely associated with
the in vivo situation (24). The observed attenuation of colony
formation indicated that GPR137 knockdown impaired the
anchorage-independent growth of gastric cancer cells. To the best
of our knowledge, the present study was the first to report that
GPR137 has a key role in mediating cancer cell growth.
Elucidating the role of GPR137 in gastric cancer
cell growth is of biological significance. GPRs have been
implicated in the mediation of cellular proliferation in diverse
types of cancer. However, GPR137 was initially identified as a GPR
in 2003 and its mRNA was only detected in brain tissues (16). Whether GPR137 functions as a key
mediator of cancer growth in the same way as other GPRs do has
remained elusive. The present study identified a novel function of
GPR137 to gastric tumorigenesis, and may provide a target for the
development of gastric cancer therapeutics.
Of note, GPR137B was previously implicated in kidney
development (14). GPR137B was
also identified to be a novel lysosome integral membrane protein
and performed certain lysosome functions, including autophagy,
degrading of products and transport of nutrition (15). However, GPR137 potentially
regulates gastric cancer cell growth via alternative mechanisms,
rather than lysosome signaling pathways. The hypothesis underlying
the present study was based upon the observation that the GPR137
transcript was detected in brain tissues, indicating that certain
ligands secreted by the brain may function as stimuli of GPR137.
The results of the present study demonstrated the tumor
promoting-effect of GPR137 in the stomach. It was therefore
suggested that specific ligands existing in the stomach and brain
may be involved in GPR137-mediated gastric cancer cell growth. A
further potential mechanism underlying the tumor-promoting effect
of GPR137 is that GPR137 may regulate molecules which are involved
in cell cycle regulation. GPR137 depletion led to abnormal
accumulation of MGC80-3 cells in the S phase and, in particular,
the G2/M phase. GPR137 may therefore contribute to
gastric cancer cell growth via manipulation of G2/M
phase regulators. A typical example of cell cycle manipulation is
that the knockdown of aurora kinase A induces G2/M phase
accumulation via the regulation of bipolar spindle formation
(25). Therefore, it was
hypothesized that GPR137 may regulate the cell cycle by influencing
G2/M phase molecules, which mediate microtubule and/or
spindle activities, and in this way promote gastric cancer cell
growth. Further study is required in order to investigate the
validity of this hypothesis.
In conclusion, to the best of our knowledge, the
present study was the first to define GPR137 as a functional
mediator of gastric cancer cell growth. Knockdown of GPR137
significantly inhibited gastric cancer cell growth in vitro.
These results may aid the development of novel gastric cancer drug
therapeutics.
References
|
1
|
Yang G, Wang Y, Zeng Y, et al: Rapid
health transition in China, 1990–2010 findings from the Global
Burden of Disease Study 2010. Lancet. 381:1987–2015. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Brenner H, Rothenbacher D and Arndt V:
Epidemiology of stomach cancer. Methods Mol Biol. 472:467–477.
2009. View Article : Google Scholar
|
|
3
|
Long N, Moore MA, Chen W, et al: Cancer
epidemiology and control in north-East Asia - past, present and
future. Asian Pac J Cancer Prev. 11(Suppl 2): S107–S148. 2010.
|
|
4
|
Li ZX and Kaminishi M: A comparison of
gastric cancer between Japan and China. Gastric Cancer. 12:52–53.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhao P, Dai M, Chen W and Li N: Cancer
trends in China. Jpn J Clin Oncol. 40:281–285. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Heng BC, Aubel D and Fussenegger M: An
overview of the diverse roles of G-protein coupled receptors
(GPCRs) in the pathophysiology of various human diseases.
Biotechnol Adv. 31:1676–1694. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dorsam RT and Gutkind JS:
G-protein-coupled receptors and cancer. Nat Rev Cancer. 7:79–94.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lappano R and Maggiolini M: G
protein-coupled receptors: novel targets for drug discovery in
cancer. Nat Rev Drug Discov. 10:47–60. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lappano R and Maggiolini M: GPCRs and
cancer. Acta Pharmacol Sin. 33:351–362. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kalyankrishna S and Grandis JR: Epidermal
growth factor receptor biology in head and neck cancer. J Clin
Oncol. 24:2666–2672. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Thomas SM, Bhola NE, Zhang Q, et al:
Cross-talk between G protein-coupled receptor and epidermal growth
factor receptor signaling pathways contributes to growth and
invasion of head and neck squamous cell carcinoma. Cancer Res.
66:11831–11839. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Arora P, Cuevas BD, Russo A, Johnson GL
and Trejo J: Persistent transactivation of EGFR and ErbB2/HER2 by
protease-activated receptor-1 promotes breast carcinoma cell
invasion. Oncogene. 27:4434–4445. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fischgräbe J, Götte M, Michels K, Kiesel L
and Wülfing P: Targeting endothelin A receptor enhances
anti-proliferative and anti-invasive effects of the HER2 antibody
trastuzumab in HER2-overexpressing breast cancer cells. Int J
Cancer. 127:696–706. 2010. View Article : Google Scholar
|
|
14
|
Spangenberg C, Winterpacht A, Zabel BU and
Löbbert RW: Cloning and characterization of a novel gene (TM7SF1)
encoding a putative seven-pass transmembrane protein that is
upregulated during kidney development. Genomics. 48:178–185. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gao J, Xia L, Lu M, et al: TM7SF1
(GPR137B): a novel lysosome integral membrane protein. Mol Biol
Rep. 39:8883–8889. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vanti WB, Nguyen T, Cheng R, Lynch KR,
George SR and O’Dowd BF: Novel human G-protein-coupled receptors.
Biochem Biophys Res Commun. 305:67–71. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee YF, Miller LD, Chan XB, et al: JMJD6
is a driver of cellular proliferation and motility and a marker of
poor prognosis in breast cancer. Breast Cancer Res. 14:R852012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sakoda T, Kasahara N, Hamamori Y and Kedes
L: A high-titer lentiviral production system mediates efficient
transduction of differentiated cells including beating cardiac
myocytes. J Mol Cell Cardiol. 31:2037–2047. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Soneoka Y, Cannon PM, Ramsdale EE, et al:
A transient three-plasmid expression system for the production of
high titer retroviral vectors. Nucleic Acids Res. 23:628–633. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tiscornia G, Singer O and Verma IM:
Production and purification of lentiviral vectors. Nat Protoc.
1:241–245. 2006. View Article : Google Scholar
|
|
21
|
Tricoli JV and Bracken RB: ZFY gene
expression and retention in human prostate adenocarcinoma. Genes
Chromosomes Cancer. 6:65–72. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Figueiredo C, Garcia-Gonzalez MA and
Machado JC: Molecular pathogenesis of gastric cancer. Helicobacter.
18(Suppl 1): S28–S33. 2013. View Article : Google Scholar
|
|
23
|
Wang D, Sun SQ, Yu YH, et al: Suppression
of SCIN inhibits human prostate cancer cell proliferation and
induces G0/G1 phase arrest. Int J Oncol. 44:161–166. 2014.
|
|
24
|
Wang LH: Molecular signaling regulating
anchorage-independent growth of cancer cells. Mt Sinai J Med.
71:361–367. 2004.PubMed/NCBI
|
|
25
|
Hata T, Furukawa T, Sunamura M, et al: RNA
interference targeting aurora kinase a suppresses tumor growth and
enhances the taxane chemosensitivity in human pancreatic cancer
cells. Cancer Res. 65:2899–2905. 2005. View Article : Google Scholar : PubMed/NCBI
|