Introduction
Hypoxia frequently accompanies various vascular
disorders, including thrombosis, atherosclerosis and
ischemia/reperfusion injury. Vascular endothelial cells
anatomically position at the interface of the blood and tissue
exchange, and therefore are frequently exposed to environments of
low oxygen tension. The two crucial functions of endothelium,
including the maintenance of a permeability barrier and the
preservation of the fluidity of blood, are adversely affected by
levels of hypoxia that occur in ischemic syndromes (1,2). The
impact of hypoxia on endothelial cells is complex and variable.
Extended durations of oxygen deprivation may result in endothelial
cell death (3–5), while moderate and brief periods of
hypoxia in these cells may activate pro-survival or proliferative
processes via the hypoxia-induced activation of signaling cascades
(6–8).
Hypoxia-inducible factor 1 (HIF-1) is a
transcription factor, which functions as a master regulator of
adaptive responses to conditions of reduced O2 (9). HIF-1 improves local microcirculation,
via its effects on vascular growth and functioning, and regulates
O2 utilization by switching oxidative metabolism to
glycolytic metabolism (10–12).
HIF-1 is composed of HIF-1α and HIF-1β subunits (13,14).
HIF-1α is the O2-regulated subunit that is specific to
HIF-1, whereas HIF-1β is also known as the aryl hydrocarbon
receptor nuclear translocator (ARNT) as it is also able to dimerize
with the aryl hydrocarbon receptor (15). Hypoxia and reoxygenation potently
increase HIF-1 transcriptional activity and HIF-1α protein
expression levels (16–19). Upregulated HIF-1α activation has
been demonstrated in the human heart under conditions of myocardial
ischemia and infarction (20), and
in patients with coronary artery disease (21,22).
Autophagy, a dynamic catabolic process in which
cellular components are delivered to the lysosome for degradation,
has been implicated in a wide array of physiological processes and
in the pathogenesis of a diverse number of diseases (23–25).
Aside from its established roles in homeostasis maintenance and
stress adaptation, autophagy also possesses functions in cellular
differentiation (26–30). It is a closely regulated process
that facilitates the maintenance of a balance among the synthesis,
degradation and subsequent recycling of cellular products. However,
it remains unclear how hypoxia-induced autophagy is involved in
hypertension and coronary disease.
The present study focuses on determining the
hypoxia-induced autophagy in human umbilical vein endothelial cells
(HUVECs) and further examining the role of autophagy on cell
viability. To the best of our knowledge, the results demonstrate
novel findings,, that autophagy is an important mechanism of
hypoxia-induced HUVEC cell viability reduction that acts via a
HIF-1-dependent pathway.
Materials and methods
Cell culture
HUVECs were cultured in M199 medium (Gibco,
Rockville, MD, USA) containing 10% fetal calf serum (Gibco) and
grown on 0.5% gelatine type A (Sigma-Aldrich, St. Louis, MO, USA)
coated T75 dishes in an incubator (5% CO2). For
augmentation, confluent HUVECs were split in 10 cm dishes using
0.25% trypsin or trypsin/EDTA. The present study was approved by
the ethics committee of Qilu Hospital of Shandong University
(Shandong, China).
Reagents and antibodies
The Beclin 1 polyclonal antibody was purchased from
Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA).
Autophagy-related gene (Atg)5 and Atg7 monoclonal antibodies were
purchased from Cell Signaling Technology Inc. (Danvers, MA, USA).
Rapamycin, 3-methyladenine (3-MA) and polyclonal antibodies for
GAPDH and LC3-II were purchased from Sigma-Aldrich (St. Louis, MO,
USA). The coding sequence of MAP1-LC3 fusion with GFP was
synthesized and cloned into the pcDNA3.1 (+) to construct the
LC3-GFP-expressing plasmid. The coding sequence of HIF-1 was
synthesized and cloned into the pcDNA3.1 (+) to construct the
HIF-1-pcDNA3.1-expressing plasmid. HIF-1 siRNA sequence was
designed and duplexes were produced by GenePharma, Co. (Shanghai,
China).
Quantitative GFP-LC3 analysis and
electron microscopy
Quantitative GFP-LC3 light microscopy autophagy
assays were performed in the HUVECs with various treatments. HUVECs
grown to 80% confluency were transfected with a GFP-LC3-expressing
plasmid, using Lipofectamine 2000 (Invitrogen Life Technologies,
Carlsbad, CA, USA). After 24 h transfection, the cells were
subjected to rapamycin treatment (200 nM) for another 24 h and
analyzed by fluorescence microscopy. In another two groups, the
cells were pretreated with hypoxia. A total of 2 h later, the cells
were subject to (3MA; 100 μM) for another 24 h and analyzed by
fluorescence microscopy. Electron microscopy was performed to
determine the autophagic vacuoles in HUVECs with or without hypoxia
treatment. Briefly, HUVEC cell samples with or without hypoxia or
rapamycin treatment, were washed three times with 1X
phosphate-buffered saline, trypsinized and collected by
centrifuging at 1,000 × g for 5 min. The cell pellets were fixed
with 4% paraformaldehyde overnight at 4°C, post-fixed with 1%
OsO4 in cacodylate buffer for 1 h at room temperature
and dehydrated stepwise with ethanol. The dehydrated pellets were
rinsed with propyleneoxide for 30 min at room temperature and then
embedded in Spurr resin for sectioning. The images of the thin
sections were observed under a transmission electron microscope
(JEM1230; JEOL, Ltd, Tokyo, Japan).
RNA isolation and quantitative
(q)PCR
Total cellular RNA from 2×105 to
5×105 cells was prepared with TRIzol and reverse
transcription (RT) was performed with M-MLV Reverse Transcriptase
(Promega Corporation, Madison, WI, USA). Primer sequences and
conditions are available upon request. For quantitative analysis of
the mRNA expression of Beclin 1, Atg5 and Atg 7, qPCR was conducted
by LightCycler 480 (Roche Applied Science, Mannheim, Germany), 1 μg
RNA per sample was converted to cDNA and used for qPCR. The data
were normalized based on GAPDH.
Plasmid overexpression and siRNA
knockdown of HIF-1
HUVECs in 6-well plates were transfected with
HIF-1-pcDNA3.1-expressing plasmid or siRNA duplexes specific for
the knockdown of human HIF-1. HIF-1 overexpression or depletion was
confirmed by western blot analysis.
Western blot analysis
The cell extracts were prepared following the
standard procedures and the proteins were detected by western
blotting using polyclonal (human) anti-Beclin 1 antibody,
monoclonal (human) anti-Atg5 antibody, monoclonal (human) anti-Atg7
antibody or polyclonal (rabbit) anti-LC3 or GAPDH antibody
(Sigma-Aldrich, St. Louis, MO, USA). Goat anti-mouse IgG or goat
anti-rabbit IgG (Pierce Biotechnology, Inc., Rockford, IL, USA)
secondary antibody conjugated to horseradish peroxidase and ECL
detection systems (Super Signal West Femto; Pierce Biotechnology,
Inc.) were used for detection.
Cell viability assay
The cell viability was determined by an MTT assay.
HUVECs were seeded in 96-well plates and following 24 h, the medium
was replaced with α-MEM medium containing 1% FBS. At 0, 12, 24 and
48 h post-treatment, the incubation medium in the test wells was
replaced with 50 μl 1X MTT solution, and the cells were incubated
for 2 h at 37°C. Following incubation, the MTT solution was
discarded and 150 μl dimethyl sulfoxide was added to dissolve the
precipitate completely at room temperature. The optical density was
then measured at 570 nm using a spectrophotometer. The cell
viability was expressed as relative viable cells (%) to control
HUVECs. All of the experiments were performed in three separate
experiments.
Statistical evaluation
For GFP-LC3 dot number analysis, the relative mRNA
expression of Beclin 1, Atg5 or Atg7 to GAPDH, MTT measurements,
statistical evaluations are presented as the mean ± standard error.
The data were analyzed using the Student’s t-test. P<0.05 was
considered to indicate a statistically significant difference
between values.
Results
Hypoxia induces autophagy and
autophagy-associated molecule expression in HUVECs
HUVECs were transfected with GFP-LC3, a biomarker
for autophagy, and exposed to hypoxic conditions for 24 h. LC3, a
microtubule-associated protein light chain 3 (MAP-LC3), typically
exhibits diffuse cytosolic distribution. Representative
fluorescence images, as demonstrated in Fig. 1A–C, indicated that the treatment of
hypoxia or rapamycin led to the redistribution of LC3 to punctate
structures and increased the number of LC3-GFP-positive vesicles in
cells. The ultrastructures of HUVECs with or without hypoxic
treatment were observed by EM microphotography (Fig. 1D–E). The prominent features of
cells subject to hypoxia treatment were observed in the form of
autophagic vacuoles and autolysosomes in the cytoplasm.
In addition, it has been demonstrated that LC3-II
increases during autophagy compared with LC3-I (31). When autophagy is activated, the
LC3-I protein localized in the cytoplasm is cleaved, lipidated and
inserted as LC3-II into autophagosome membranes. To detect the
expression of LC3-II, western blotting was performed with lysates
from HUVECs subject to hypoxia or rapamycin (Fig. 2A). As part of a type III PI3 kinase
complex, the autophagy gene Beclin 1, which is required for the
formation of the autophagic vesicles (32). Also, autophagy-related gene (ATG)
products have essential roles in autophagy, such as Atg 5 and Atg
7. Fig. 2B and C demonstrates that
the expression of Atg 5, Atg 7 and Beclin 1 at an mRNA and protein
level increased following hypoxia or rapamycin treatment. These
results significantly indicate that hypoxia or rapamycin treatment
induces autophagy in HUVECs.
Hypoxia-induced autophagy-related
molecule expression in HUVECs may be regulated by HIF-1
To reveal the precise underlying mechanisms behind
the above demonstrated activation of autophagy in HUVECs, the
alteration of HIF-1 expression with a pcDNA3.1 eukaryotic plasmid
and chemical synthesized siRNA was further examined. HIF-1, a key
transcription factor, has pivotal roles in hypoxia. The HIF-1
overexpression with eukaryotic plasmid or HIF-1 knockout by siRNA
was transfected to the HUVECs. As revealed in Fig. 3A–D, overexpression of HIF-1
significantly improved the occurrence of autophagy, and HIF-1
knockout attenuated the formation of autophagic vacuoles in HUVEC
cells. More importantly, HIF-1 overexpression markedly promoted the
cleavage of LC3 and expression of Atg5, Atg7 and Beclin1. By
contrast, transfection with HIF-1 siRNA, which respectively blocked
HIF-1 expression, significantly prevented LC3-II production, as
well as the expression of Atg5, Atg7 and Beclin 1 (Fig. 3E and F). Therefore, these data
demonstrated that autophagy to hypoxia is dependent on HIF-1 in
HUVEC cells. It may therefore be concluded that the HIF-1 pathway
regulates autophagy activation in HUVECs under hypoxic
conditions.
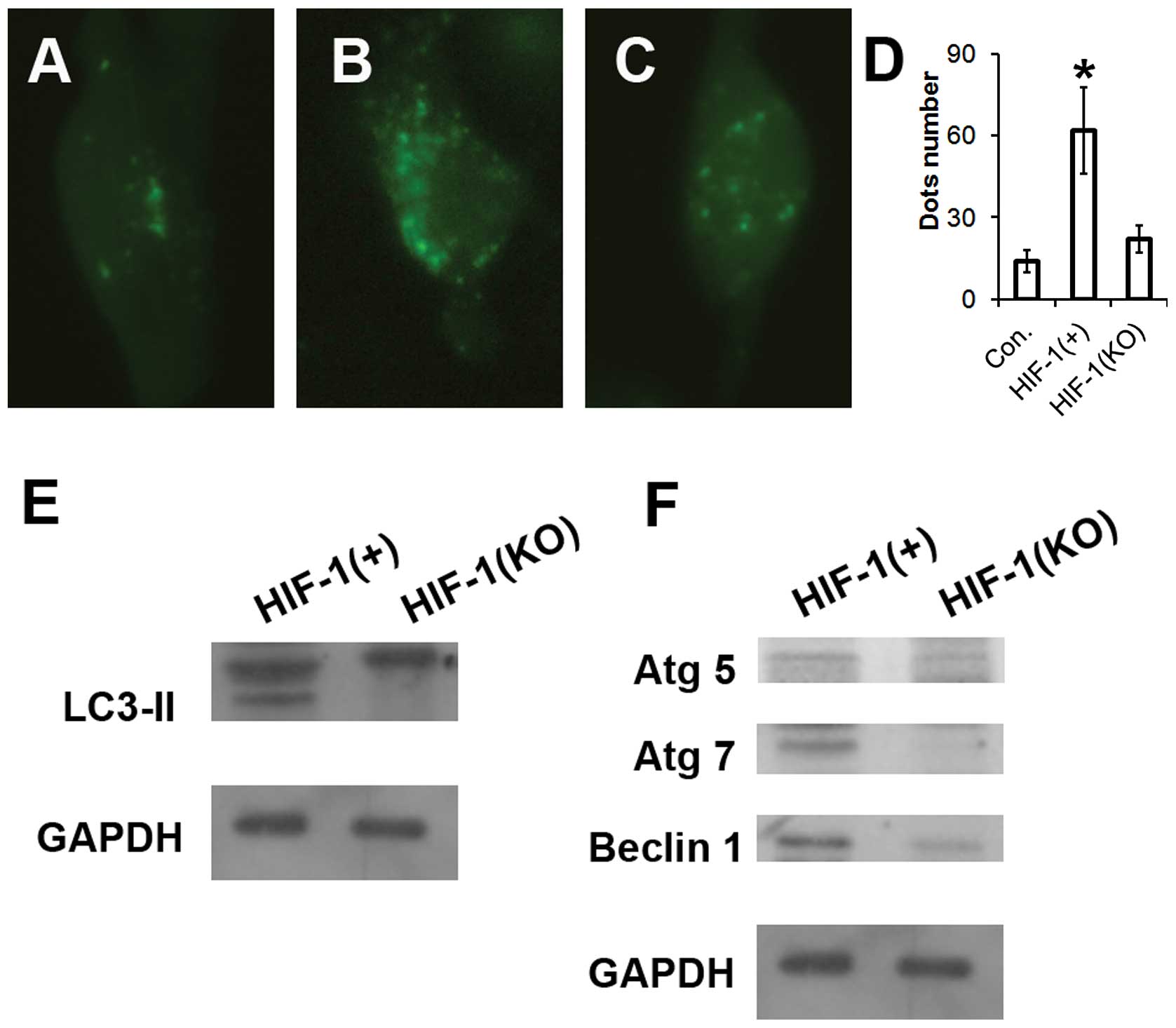 | Figure 3Hypoxia induces autophagy in HUVECs
through the HIF-1 pathway. (A, B, C and D) HUVEC cells were
transfected with a plasmid that expresses a GFP-LC3 fusion protein.
Following 24 h, the cells were incubated for another 24 h at 37°C
in Dulbecco’s modified Eagle’s medium with 1/10,000 dimethyl
sulfide (control), pcDNA-HIF-1 or HIF-1 siRNA as described in the
Materials and methods. Following fixation, the cells were
immediately visualized by fluorescence microscopy. The number of
punctate GFP-LC3 dots in each cell was counted and at least 100
cells were included for each group. *P<0.01 vs.
control. (E and F) Following pcDNA-HIF-1 or HIF-1 siRNA incubation
for 24 h, as described in the Materials and methods, the cells were
lysed and subjected to western blotting with the antibodies
indicated. HIF-1, hypoxia-inducible factor 1; HUVECs, human
umbilical vein endothelial cells. |
Decrease in HUVEC cell viability due to
hypoxia-induced autophagy is dependent on HIF-1
As the results above indicated, HIF-1 induced
significant autophagy and a high level expression of
autophagy-associated molecules in HUVECs. Firstly, western blotting
was performed to examine the expression of HIF-1 or LC3-II induced
by hypoxic conditions or HIF-1 transfection with a pcDNA plasmid in
HUVECs. As revealed in Fig. 4A–B,
the expression of HIF-1 or LC3-II protein stimulated by HIF-1
transfection with a pcDNA plasmid was notably higher than that with
hypoxia condition. To further determine the affect of HIF-1 or
hypoxia induced autophagy on the cell viability, an MTT assay was
conducted to reveal the HUVEC cell viability. It was demonstrated
that the cells treated with HIF-1 or hypoxia decreased
significantly in viability (Fig.
4C). In addition, HIF-1 reduced the cell viability more than
hypoxia in the HUVEC cells. Finally, to further confirm the
functional significance of HIF-1 in hypoxia-induced cell viability,
the HUVEC cells were pretreated with the pharmacological inhibitor
of autophagy, 3-MA, followed by exposure to hypoxia. The data
revealed that 3-MA treatment evidently prevented viability
decreasing in HUVEC cells when cultured under hypoxic conditions
(Fig. 4D). In conclusion, the
above data suggests that HIF-1 indeed has an important role in the
hypoxia-induced reduction in cell viability.
 | Figure 4Autophagy induced by high dose of
HIF-1 expression reduced the HUVEC cell viability. (A and B)
Following pcDNA3.1, pcDNA-HIF-1 transfection or hypoxia treatment
of 24 h, as described in the Materials and methods, the cells were
lysed and subjected to western blotting with the antibodies
indicated. Densitometry was performed for quantification. (C)
Viability of HUVEC cells post treatment with pcDNA3.1, pcDNA-HIF-1
transfection or hypoxia treatment. HUVEC cells 12, 24 or 48 h post
inoculation with pcDNA3.1, pcDNA-HIF-1 transfection or hypoxia
treatment decreased significantly in viability.
*P<0.05. (D) 3MA may ameliorate the hypoxia-induced
HUVEC cell viability decrease, *P<0.05 vs. pcDNA3.1.
HIF-1, hypoxia-inducible factor 1; HUVECs, human umbilical vein
endothelial cells; 3MA, 3-Methyladenine. |
Discussion
Previous studies have documented that
hypoxia-induced cell death is a major concern clinically and has a
critical role in various physiological processes, including cell
proliferation, hypoxic/ischaemic disease, organ transplantation,
angiogenesis, tumor invasion and metastasis (33–36).
Clinical studies have demonstrated that autophagy is an
intracellular lysosomal degradation process that is characterized
by the formation of double-membrane vesicles in cytoplasm.
Therefore, autophagy has key physiological cellular functions,
including degradation of long-lived proteins, organelle turnover,
adaptation to nutrient depletion, extension of lifespan, cellular
development, differentiation and anti-aging (37). Consequently, autophagy may preserve
cell viability or be a self-destructive process that leads to cell
death (38).
In the present study, an increased in autophagic
activity in HUVECs induced by hypoxia was detected. HUVECs are
widely used as a source of vascular endothelial cells. To the best
of our knowledge, this is the first study to report the effects of
hypoxia on autophagic cell death in HUVEC cells. Several approaches
were adopted to determine autophagy, including fluorescent LC3-GFP
dots, electron microscopic imaging and autophagy-associated
molecules expression in mRNA and protein levels. HUVEC cells were
exposed to hypoxia and an evident increase in the number of
autophagic vacuoles and autolysosomes in the cytoplasm was
identified, suggesting enhanced autophagy formation. In addition,
the occurrence of autophagy was also confirmed with rapamycin
treatment (Fig. 1). The
determination of further autophagy-associated molecules also
confirmed the autophagy induced by hypoxia or rapamycin, as
evidenced by the increase in the expression of LC3-II following the
treatments, aswell as the enhanced expression of Beclin 1, Atg 5
and Atg 7 in the HUVEC cells post hypoxia or rapamycin treatment at
mRNA and protein levels (Fig. 2).
In conclusion, autophagy may be induced by hypoxia in HUVEC cells
in vitro.
HIF-1 is a transcription factor that is essential in
the regulation of gene expression to maintain oxygen homeostasis
(39). Firstly, HIF-1 was proved
to coordinate adaptive responses to hypoxia at the cellular and
systemic levels (14,40,41).
Furthermore, HIF-1 has been demonstrated to regulate the expression
of hundreds of target genes involved in angiogenesis,
erythropoiesis, metabolism, autophagy and other adaptive responses
to hypoxia (42). HIF is a
heterodimer of an α subunit that is unstable in the presence of
relatively high levels of oxygen and a β-subunit that is not oxygen
regulated. To elucidate the mechanisms by which HUVEC cells respond
to hypoxia, an HIF-1 overexpression plasmid and HIF-1 siRNA were
used and it was demonstrated that hypoxia triggered autophagy in
HUVEC cells via the HIF-1 pathway. A significant increase in the
autophagy-specific autolysosomes was observed via the LC3-GFP
report vector with a fluorescence microscope in HIF-1-treated
cells. Coversely, HIF-1 knockout with siRNA treatment in HUVEC
cells led to a notable reduction in autolysosome formation. In
addition, the cleavage and recruitment of LC3 to autophagosomes, as
well as the expression of autophagy-associated molecules were also
enhanced by HIF-1 overexpression and attenuated by HIF-1 knockout
(Fig. 3). In conclusion,
hypoxia-induced autophagy in HUVEC cells was demonstrated to
involve the HIF-1 pathway.
Hypoxia treatment or HIF-1 overexpression may induce
autophagy, however, their effects on cell viability appeared to be
different. Hypoxia appeared to ameliorate the cell viability;
however, cell viability was more markedly decrease following HIF-1
stimulation. HIF-1 was able to significantly inhibit the HUVEC cell
viability and this effect was reversed by 3MA (Fig. 4). Therefore, the autophagy induced
by HIF-1 was able to destroy cellular components and acted as a
self-destructive process that led to cell death. It may therefore
be a novel mechanism involved in the decreased proliferation of the
HUVEC cells post treatment with HIF-1.
In summary, the present study suggests that hypoxia
induces autophagy in HUVEC cells, due to activation of the HIF-1
pathway. HIF-1 treatment was able to induce excess autophagy and
reduce the cell viability. Further studies are required to
elucidate the mechanisms of hypoxia-induced apoptosis and
autophagic cell death, which may facilitate the development of
novel methods for the prevention of hypoxic-ischaemic diseases.
Acknowledgements
This study was supported by the grant from Qilu
Hospital of Shandong University (Shandong, China).
References
|
1
|
Stelzner TJ, O’Brien RF, Sato K and Weil
JV: Hypoxia-induced increases in pulmonary transvascular protein
escape in rats. Modulation by glucocorticoids. J Clin Invest.
82:1840–1847. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hamer JD, Malone PC and Silver IA: The
PO2 in venous valve pockets: its possible bearing on
thrombogenesis. Br J Surg. 68:166–170. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kloner RA, Rude RE, Carlson N, et al:
Ultrastructural evidence of microvascular damage and myocardial
cell injury after coronary artery occlusion: which comes first?
Circulation. 62:945–952. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lee C, Cheng W, Chang M, et al:
Hypoxia-induced apoptosis in endothelial cells and embryonic stem
cells. Apoptosis. 10:887–894. 2005. View Article : Google Scholar
|
|
5
|
Stempien-Otero A, Karsan A, Cornejo CJ, et
al: Mechanisms of hypoxia-induced endothelial cell death. Role of
p53 in apoptosis. J Biol Chem. 274:8039–8045. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee S, Chen TT, Barber CL, et al:
Autocrine VEGF signaling is required for vascular homeostasis.
Cell. 130:691–703. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mogi M, Fukuo K, Yang J, Suhara T and
Ogihara T: Hypoxia stimulates release of the soluble form of fas
ligand that inhibits endothelial cell apoptosis. Lab Invest.
81:177–184. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schäfer M, Schäfer C, Ewald N, Piper HM
and Noll T: Role of redox signaling in the autonomous proliferative
response of endothelial cells to hypoxia. Circ Res. 92:1010–1015.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Semenza GL: Hypoxia-inducible factors in
physiology and medicine. Cell. 148:399–408. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shohet RV and Garcia JA: Keeping the
engine primed: HIF factors as key regulators of cardiac metabolism
and angiogenesis during ischemia. J Mol Med (Berl). 85:1309–1315.
2007. View Article : Google Scholar
|
|
11
|
Taylor CT: Mitochondria and cellular
oxygen sensing in the HIF pathway. Biochem J. 409:19–26. 2008.
View Article : Google Scholar
|
|
12
|
Semenza GL: Hypoxia-inducible factor 1:
regulator of mitochondrial metabolism and mediator of ischemic
preconditioning. Biochim Biophys Acta. 1813:1263–1268. 2011.
View Article : Google Scholar
|
|
13
|
Wang GL and Semenza GL: Purification and
characterization of hypoxia-inducible factor 1. J Biol Chem.
270:1230–1237. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang GL, Jiang BH, Rue EA and Semenza GL:
Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS
heterodimer regulated by cellular O2 tension. Proc Natl
Acad Sci USA. 92:5510–5514. 1995. View Article : Google Scholar
|
|
15
|
Reyes H, Reisz-Porszasz S and Hankinson O:
Identification of the Ah receptor nuclear translocator protein
(Arnt) as a component of the DNA binding form of the Ah receptor.
Science. 256:1193–1195. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cai Z, Manalo DJ, Wei G, et al: Hearts
from rodents exposed to intermittent hypoxia or erythropoietin are
protected against ischemia-reperfusion injury. Circulation.
108:79–85. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yuan G, Nanduri J, Bhasker CR, Semenza GL
and Prabhakar NR: Ca2+/calmodulin kinase-dependent
activation of hypoxia inducible factor 1 transcriptional activity
in cells subjected to intermittent hypoxia. J Biol Chem.
280:4321–4328. 2005. View Article : Google Scholar
|
|
18
|
Yuan G, Nanduri J, Khan S, Semenza GL and
Prabhakar NR: Induction of HIF-1alpha expression by intermittent
hypoxia: involvement of NADPH oxidase, Ca2+ signaling,
prolyl hydroxylases, and mTOR. J Cell Physiol. 217:674–685. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Peng YJ, Yuan G, Ramakrishnan D, et al:
Heterozygous HIF-1alpha deficiency impairs carotid body-mediated
systemic responses and reactive oxygen species generation in mice
exposed to intermittent hypoxia. J Physiol. 577:705–716. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee SH, Wolf PL, Escudero R, et al: Early
expression of angiogenesis factors in acute myocardial ischemia and
infarction. N Engl J Med. 342:626–633. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hlatky MA, Quertermous T, Boothroyd DB, et
al: Polymorphisms in hypoxia inducible factor 1 and the initial
clinical presentation of coronary disease. American Heart J.
154:1035–1042. 2007. View Article : Google Scholar
|
|
22
|
Resar JR, Roguin A, Voner J, et al:
Hypoxia-inducible factor 1alpha polymorphism and coronary
collaterals in patients with ischemic heart disease. Chest.
128:787–791. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shintani T and Klionsky DJ: Autophagy in
health and disease: a double-edged sword. Science. 306:990–995.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rosenfeldt MT and Ryan KM: The role of
autophagy in tumour development and cancer therapy. Expert Rev Mol
Med. 11:e362009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Todde V, Veenhuis M and van der Klei IJ:
Autophagy: principles and significance in health and disease.
Biochim Biophys Acta. 1792:3–13. 2009. View Article : Google Scholar
|
|
26
|
Boya P, Mellén MA and de la Rosa EJ: How
autophagy is related to programmed cell death during the
development of the nervous system. Biochem Soc Trans. 36:813–817.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kundu M, Lindsten T, Yang CY, et al: Ulk1
plays a critical role in the autophagic clearance of mitochondria
and ribosomes during reticulocyte maturation. Blood. 112:1493–1502.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pua HH, Guo J, Komatsu M and He YW:
Autophagy Is Essential for Mitochondrial Clearance in Mature T
Lymphocytes. JJ Immunol. 182:4046–4055. 2009. View Article : Google Scholar
|
|
29
|
Singh R, Xiang Y, Wang Y, et al: Autophagy
regulates adipose mass and differentiation in mice. J Clin Invest.
119:3329–3339. 2009.PubMed/NCBI
|
|
30
|
Srinivas V, Bohensky J and Shapiro IM:
Autophagy: a new phase in the maturation of growth plate
chondrocytes is regulated by HIF, mTOR and AMP kinase. Cells
Tissues Organs. 189:88–92. 2009. View Article : Google Scholar :
|
|
31
|
Kabeya Y, Mizushima N, Ueno T, et al: LC3,
a mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sun Y and Peng ZL: Programmed cell death
and cancer. Postgraduate Med J. 85:134–140. 2009. View Article : Google Scholar
|
|
33
|
Volm M and Koomägi R: Hypoxia-inducible
factor (HIF-1) and its relationship to apoptosis and proliferation
in lung cancer. Anticancer Res. 20:1527–1533. 2000.PubMed/NCBI
|
|
34
|
Rayner BS, Duong TT, Myers SJ and Witting
PK: Protective effect of a synthetic anti-oxidant on neuronal cell
apoptosis resulting from experimental hypoxia re-oxygenation
injury. J Neurochem. 97:211–221. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bhogal RH, Weston CJ, Curbishley SM, et
al: Variable responses of small and large human hepatocytes to
hypoxia and hypoxia/reoxygenation (H-R). FEBS Lett. 585:935–941.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Härtel FV, Holl M, Arshad M, et al:
Transient hypoxia induces ERK-dependent anti-apoptotic cell
survival in endothelial cells. American J Physiol Cell Physiol.
298:C1501–C1509. 2010. View Article : Google Scholar
|
|
37
|
Mazure NM and Pouysségur J:
Hypoxia-induced autophagy: cell death or cell survival? Curr Opin
Cell Biol. 22:177–180. 2010. View Article : Google Scholar
|
|
38
|
Tsujimoto Y and Shimizu S: Another way to
die: autophagic programmed cell death. Cell Death Differ. 12(Suppl
2): 1528–1534. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Semenza GL: HIF-1 and mechanisms of
hypoxia sensing. Curr Opin Cell Biol. 13:167–171. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Iyer NV, Kotch LE, Agani F, et al:
Cellular and developmental control of O2 homeostasis by
hypoxia-inducible factor 1 alpha. Genes Dev. 12:149–162. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yu AY, Shimoda LA, Iyer NV, et al:
Impaired physiological responses to chronic hypoxia in mice
partially deficient for hypoxia-inducible factor 1alpha. J Clin
Invest. 103:691–696. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
42
|
Semenza GL: Regulation of oxygen
homeostasis by hypoxia-inducible factor 1. Physiology. 24:97–106.
2009. View Article : Google Scholar : PubMed/NCBI
|
















