Introduction
Numerous radionuclides used in the diagnosis and
therapy of disease are β particle emitters (153Sm-EDTMP,
Na131I, 186Re-HEDP and 89SrCl) and
demonstrate promising therapeutic results. To date, a large number
of preclinical and clinical studies have been performed on emitting
radionuclides (1,2). Iodine-131 is known to destroy
residual thyroid tissue following surgical resection of
differentiated thyroid carcinoma (3) and is widely used to treat
hyperthyroidism (4). In addition,
iodine-131 has been conjugated to antibody derivatives and used to
treat other malignant diseases by radioimmunotherapy (RIT). Cell
death induced by RIT has been demonstrated to trigger the apoptosis
of tumor cells (5). The cytotoxic
effect of iodine-131 on the thyroid cancer cell line, BCPAP, has
been investigated and low iodine-131 doses have been found to
result in cell apoptosis (6).
However, the mechanism through which iodine-131 induces cell death
in the thyrocyte cell line, HTori-3, remains to be fully
elucidated.
Radiation-induced DNA damage promotes the expression
of the p53 gene at the post-translational level (7). Activation of p53 leads to cell growth
arrest at G1 and/or G2 phase, DNA repair, senescence or apoptosis
(8,9). The B-cell lymphoma 2 (Bcl-2) family
is divided into proapoptotic proteins, including Bcl-2 associated X
protein (Bax), and antiapoptotic proteins, including Bcl-2
(10). Whether a cell undergoes
apoptosis depends, to a certain extent, on the ratio of Bcl-2 to
Bax (11). The cell-surface
receptor, Fas [also known as CD95 or apoptosis antigen-1 (Apo-1)],
is a key component of the extrinsic death pathway and is activated
through the binding of its ligand, FasL (12). Radiation-induced cell cycle arrest
at the G1 and G2 restriction points allows cells to repair DNA
damage prior to DNA synthesis and cell division (13,14).
A number of thyroid cancer cell lines have been
demonstrated to lose the expression of thyroid-specific genes and
have altered karyotypes, including inactivation of the tumor
suppressor gene, p53 (15).
HTori-3 cell line is a nontumorigenic, immortalized human thyrocyte
cell line and preserves the most specialized functions of
differentiated thyroid cells (16). In the present study, the HTori-3
cell line was used as a model and the cytotoxic effect of
iodine-131 on the human thyrocyte cell line, HTori-3, was
investigated. In addition, the possible underlying mechanisms of
iodine-131-induced cell apoptosis, cell cycle arrest and the effect
of iodine-131 therapy in hyperthyroidism were assessed, including
the p53 pathway.
Materials and methods
Cell culture
The HTori-3 cell line was provided by Professor Peng
Hou (Endocrinology Laboratory, The First Affiliated Hospital of
Xi’an Jiaotong University College of Medicine, Xi’an, China). The
cells were cultured in RMPI-1640 medium (Hyclone, Logan, UT, USA)
and supplemented with 10% heat-inactivated fetal bovine serum
(Minhai, Lanzhou, China) and 1% penicillin/streptomycin (Hyclone)
in a 5% CO2 humidified atmosphere at 37°C. The study was
approved by the ethics committee of the First Affiliated Hospital
of Xi’an Jiaotong University, Xi’an, China.
Iodine-131 uptake assay
The cells were seeded at 1×105/well in
6-well plates for 24 h. Subsequently, the cells were cultured for
24 h with 2 ml culture medium per well containing 14.8 MBq/ml
iodine-131. Next, the iodine-131-containing medium was decanted,
and the cells were washed twice with phosphate-buffered saline
(PBS) and trypsinized (Hyclone) to count the total cell number. The
radioactivity of the cells was measured with a radioactivity
counter (Sim-max LSA3000; Sim-max Technology Co., Ltd, Shanghai,
China).
Cell viability assay
Cell viability was determined using an MTT assay
[3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; MP
Biomedicals, Santa Ana, CA, USA]. The cells were seeded at
1.5×104/ml in 96-well plates for 24 h. Subsequently,
iodine-131 was added to achieve final iodine-131 activity
concentrations of 7.4, 14.8 and 29.6 MBq/ml. Next, the cells were
exposed to iodine-131 treatment for 12, 24, 48, 72 and 96 h with
six replicates for each concentration value, while identical
volumes of PBS were added to the control wells. Following addition
of 40 μl/well MTT, the samples were incubated for a further 4 h.
The medium was discarded and the formazan crystals were solubilized
in 150 μl/well DMSO. The sample absorbance at 570 nm/630 nm was
measured using a Multiskan plate reader (BioTek Instruments, Inc.,
Winooski, VT, USA). The absorbance was directly associated with the
viable cell number. The experiment was performed at least in
triplicate.
Cell apoptosis assay
A cell apoptosis assay was performed using an
Annexin-V-fluorescein isothiocyanate (FLOUS) staining kit (Roche
Diagnostics GmbH, Mannheim, Germany). The cells were seeded at
5×104/ml in 6-well plates for 24 h and then treated with
various doses of iodine-131 (7.4, 14.8 or 22.2 MBq/ml) (2) for 48 h or 14.8 MBq/ml iodine-131 for
different time periods (24, 48 or 72 h). A dose of 14.8 MBq/ml
iodine-131 was selected for evaluation over various time-points
following preliminary experiments which revealed low apoptotic
levels with a dose of 7.4 MBq/ml and almost total apoptosis with a
dose of 22.2 MBq/ml. At the designated time-points, the medium and
the trypsinized cells were collected in a centrifuge tube.
Subsequently, the cells were washed with PBS and centrifuged at 200
× g for 5 min. Finally, the cells were resuspended in 100 μl of
Annexin-V-FLOUS labeling solution and analyzed by flow cytometry
(BD Biosciences, San Jose, CA, USA).
Cell cycle assay
The cells were seeded at 1×105/well in
6-well plates for 24 h and then exposed to various iodine-131 doses
(7.4, 14.8 or 22.2 MBq/ml) for a further 24 h or the same dose
(14.8 MBq/ml) for different time periods (12, 24 and 48 h). At the
various time points, the cells were trypsinized and fixed in
ice-cold 70% ethanol, followed by storing at 4°C overnight.
Following centrifugation at 1,000 rpm for 5 min, the samples were
resuspended in a solution containing 50 g/ml propidium iodide (MP
Biomedicals) and 100 U/ml RNase (MP Biomedicals). The DNA content
of the nuclei was measured using flow cytometry (BD Biosciences),
counting 15,000 gated events.
Western blot analysis
HTori-3 cells were treated with 14.8 MBq/ml
iodine-131 for different time periods (12, 24 and 48 h). At the
indicated times following irradiation, total protein was extracted
using the radioimmunoprecipitation assay buffer (Wolsen, Xi’an,
China). Total protein (~150 μg) was denatured in loading buffer at
100°C for 10 min and electrophoresed using 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Lysates
containing 150 μg proteins were subjected to SDS-PAGE, followed by
transfer of proteins to a polyvinylidene difluoride membrane (EMD
Millipore, Billerica, MA, USA). The membranes were blocked with 5%
bovine serum albumin (MP Biomedicals LLC, Santa Ana, CA, USA) for 1
h at room temperature and incubated overnight at 4°C with the
primary rabbit anti-human p53, mouse anti-human Bcl-2 and mouse
anti-human Fas monoclonal antibodies (1:1,000; Epitomics,
Burlingame, CA, USA). Subsequently, the membranes were washed with
Tris-buffered saline containing 0.1% Tween-20 for 20–30 min,
followed by incubation with the secondary antibody (1:20). The
secondary antibodies used were horseradish peroxidase-labeled
anti-rabbit or -mouse immunoglobulin (Ig)G (Santa Cruz
Biotechnology, Heidelberg, Germany). Membranes reprobed with mouse
anti-GAPDH (Abmart, Arlington, MA, USA) were used as the internal
control for equal protein loading.
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
The RNA was extracted from cells with TRIzol reagent
(Takara Biotechnology Co., Ltd., Dalian, China) according to the
manufacturer’s instructions. The concentration of total RNA was
determined using a spectrophotometer (BioTek Instruments, Inc.,
Winooski, VT, USA) and the 260/280 absorbance ratio, used to
investigate the sample purity, was between 1.8 and 2.0. cDNA
synthesis was performed using 500 ng total RNA in a reaction volume
of 10 μl. The thermal cycling conditions were as follows: 15 min at
37°C and 5 sec at 85°C. Copy number quantification was performed
with RT-qPCR using β-actin as the internal reference. The primer
sequences of p53, Bcl-2, Fas, growth arrest and DNA
damage-inducible 45 (GADD45) and β-actin are shown in Table I. Following reverse transcription,
the conditions were as follows: 95°C for 2 min, 40 cycles of 15 sec
at 95°C and 60 sec at 60°C for annealing and extension were run on
a CFX96 Thermal Cycler Dice™ real-time PCR system (Bio-Rad
Laboroatories, Inc., Hercules, CA, USA). The RT-qPCR reaction was
conducted with 100 ng cDNA in a 20 μl reaction volume containing 10
μl SYBR® Premix Ex Taq™ II (Takara Biotechnology Co.,
Ltd.), 0.4 μM forward primer, 0.4 μM reverse primer and 6.4 μl
dH2O. The relative representation of the target gene
copy number in the irradiated cells with respect to the
non-irradiated control was obtained using the formula
2−ΔΔCt, which is a simplified calculation tool derived
from the ΔΔCt method for gene expression analysis. Each sample was
analyzed twice and the mean values were calculated for statistical
analysis.
 | Table IPrimers sequences of p53, Bcl-2, Fas,
GADD45 and β-actin. |
Table I
Primers sequences of p53, Bcl-2, Fas,
GADD45 and β-actin.
| Gene | Forward | Reverse | Length (bp) |
|---|
| p53 |
CTCCTCAGCATCTTACCGAGT |
GCTGTTCCGTCCCAGTAGATTA | 239 |
| Bcl-2 |
ATGTGTTGGAGAGCGTCAAC |
AGAGACAGCCAGGAGAAATCAAAC | 182 |
| Fas |
TGGCTTTGTCTTCTTCTTTTGC |
CTTGGTTTTCCTTTCTGTGCTT | 97 |
| GADD45 |
CCCAGTGGACAGCGAGCCAGC |
ACTGCAGGCTTCCTGTGGGC | 137 |
| β-actin | CTGGGACATGGAGAAA | AAGGAAGGAAGAGTGC | 285 |
Statistical analysis
Data are presented as the mean ± standard deviation.
Statistical analyses were performed using SPSS 16.0 statistical
software for Windows (SPSS, Inc., Chicago, IL, USA). The
statistical significance of differences between the groups were
investigated using Student’s unpaired t-test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Human thyrocyte HTori-3 cells exhibit
iodine uptake activity
To determine whether the HTori-3 cells possessed
iodine uptake activity, the iodine uptake was assessed with or
without iodine-131 treatment. Following treatment with 14.8 MBq/ml
iodine-131 for 24 h, the radioactivity count was found to be
51128±6.02 cpm, while the radioactivity in the control group was
366.5±4.13 cpm, indicating that HTori-3 cells possessed iodine
uptake activity.
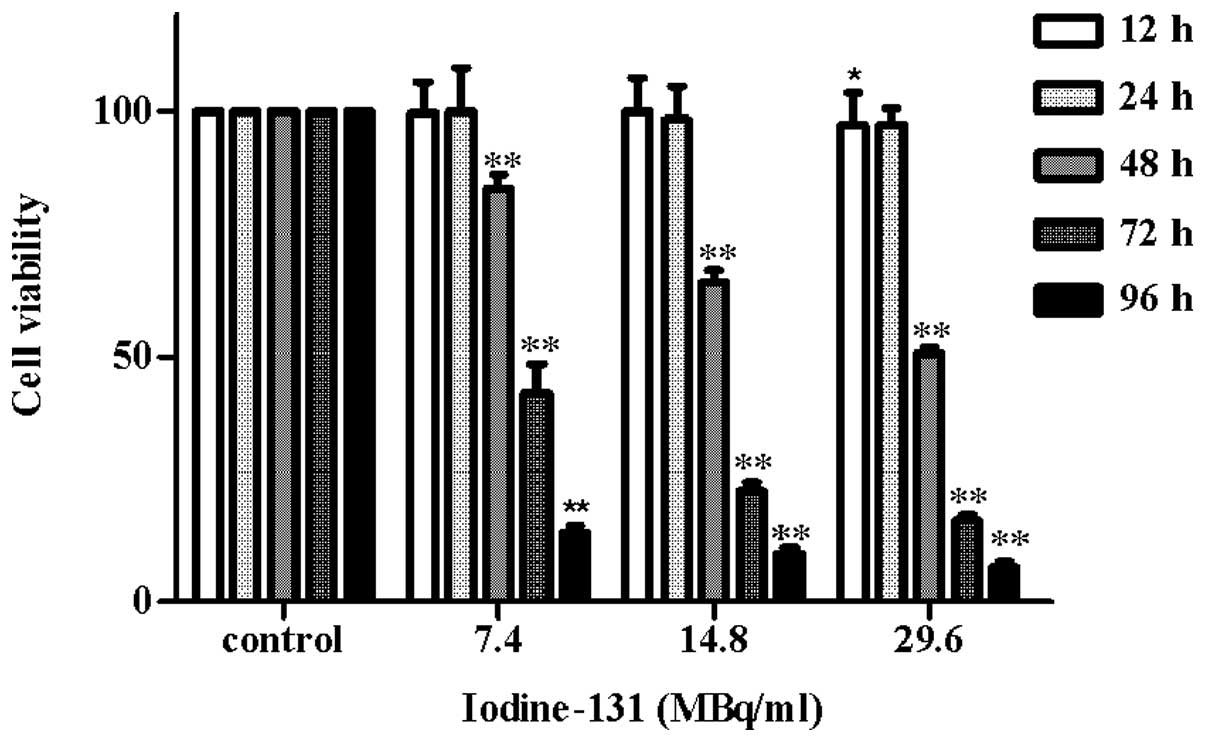
Iodine-131 inhibits cell growth in a
time- and dose-dependent manner
In order to investigate the effect of iodine-131 on
human thyrocyte cells, HTori-3 cells were incubated with various
concentrations of iodine-131 (7.4, 14.8 or 29.6 MBq/ml) for
designated time periods (12, 24, 48, 72 and 96 h) and the cell
viability was determined using an MTT assay. The results (Fig. 1) revealed that iodine-131 produced
a significant inhibition of cell growth compared with the control
group. Notably, 15.7% inhibition of cell growth was observed at a
dose of 7.4 MBq/ml (P<0.01) and 49.5% inhibition was observed at
a dose of 29.6 MBq/ml (P<0.01) after 48 h. Furthermore,
treatment with 14.8 MBq/ml iodine-131 induced a significant
reduction in cell viability (34.8%) after 48 h (P<0.01), which
was maintained to 93.2% after 96 h (P<0.01). The experiments
revealed that iodine-131 exerted a cytotoxic effect on the human
thyrocyte cell line. Growth inhibition was induced in a time- and
dose-dependent manner. The iodine-131 dose required for 50% growth
inhibition of HTori-3 cells after 48 h was 27.75±2.22 MBq/ml.
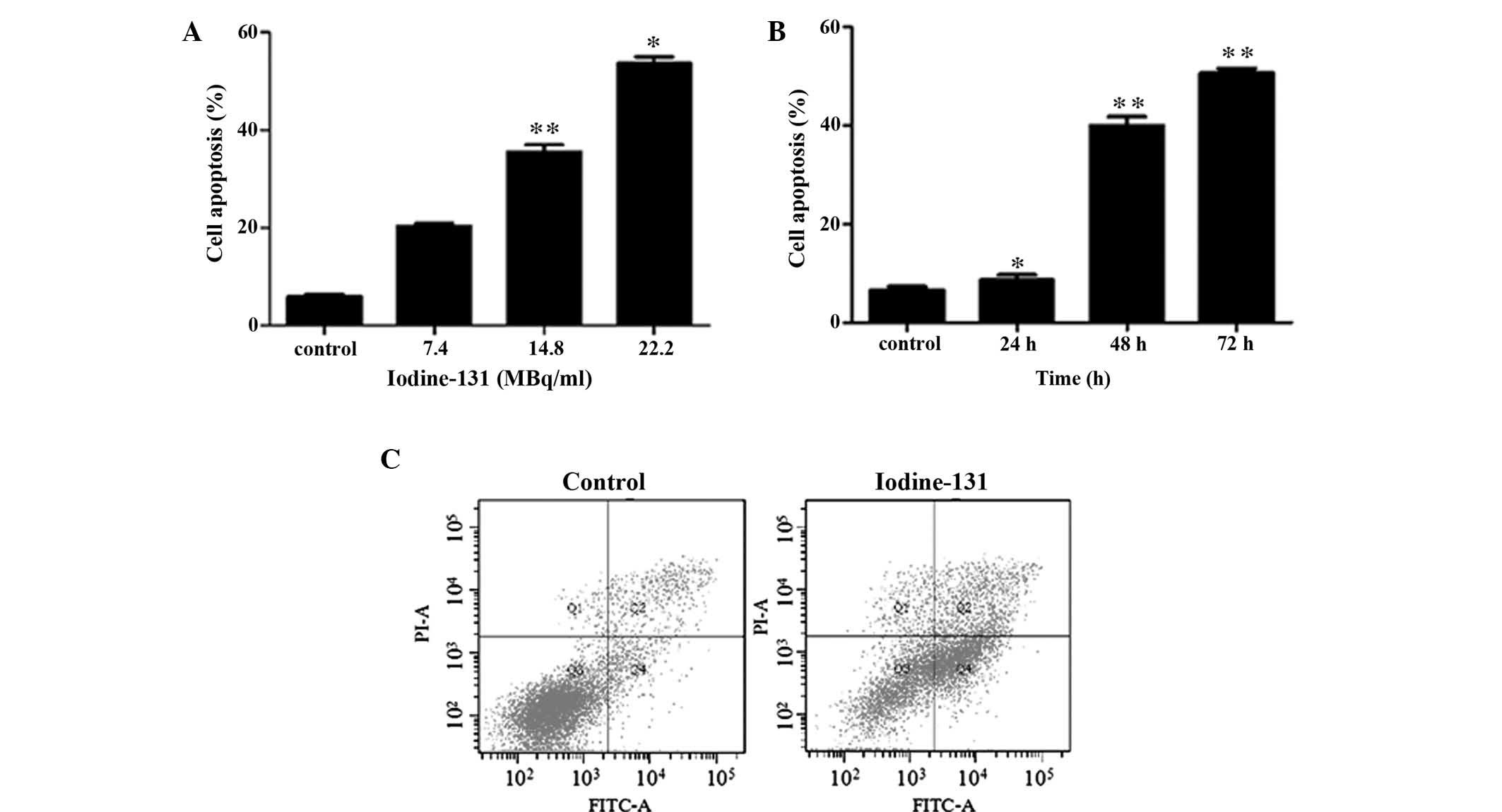
Iodine-131 induces growth inhibition of
HTori-3 cells via cell cycle arrest and apoptosis
To determine whether iodine-131 affected the HTori-3
cell apoptotic response, HTori-3 cells were treated with different
doses of iodine-131 (7.4, 14.8 or 22.2 MBq/ml) for 48 h or with
14.8 MBq/ml iodine-131 for various time periods (24, 48 or 72 h).
Apoptosis was analyzed using flow cytometry. The negative control
was treated with PBS. Apoptosis was observed in the HTori-3 cells
upon iodine-131 administration in a time- and dose-dependent
manner. The apoptotic cell percentage 48 h after iodine-131
treatment was 20.3% at 7.4 MBq/ml, 35.4% at 14.8 MBq/ml (P<0.01)
and 53.6% at 22.2 MBq/ml (P<0.05), while only 5.8% cell death
via apoptosis was observed in the control group (Fig. 2A). Evident apoptosis was induced at
48 h after treatment with 14.8 MBq/ml iodine-131 (39.9%;
P<0.01), while a higher number of apoptotic cells was observed
at 72 h (50.6%; P<0.01; Fig.
2B).
The capacity of iodine-131 to inhibit cell cycle
progression was evaluated via flow cytometry. A representative
example depicting the effect of iodine-131 treatment on cell cycle
phase distribution in the HTori-3 cell line is shown in Fig. 3. At 24 after iodine-131 treatment,
the percentages of cells in the G2/M phase were 24.4% at 7.4 MBq/ml
(P<0.05), 38.6% at 14.8 MBq/ml (P<0.01) and 72.4% at 22.2
MBq/ml (P<0.05), while the percentage of the control group was
13.5%. Treatment with 14.8 MBq/ml iodine-131 for 24 h resulted in
an accumulation of 39.2% cells in the G2/M phase compared with the
control (19.1%; P<0.01). The G2/M accumulation was more
pronounced in cells treated with iodine-131 for 48 h (42.3%;
P<0.01).
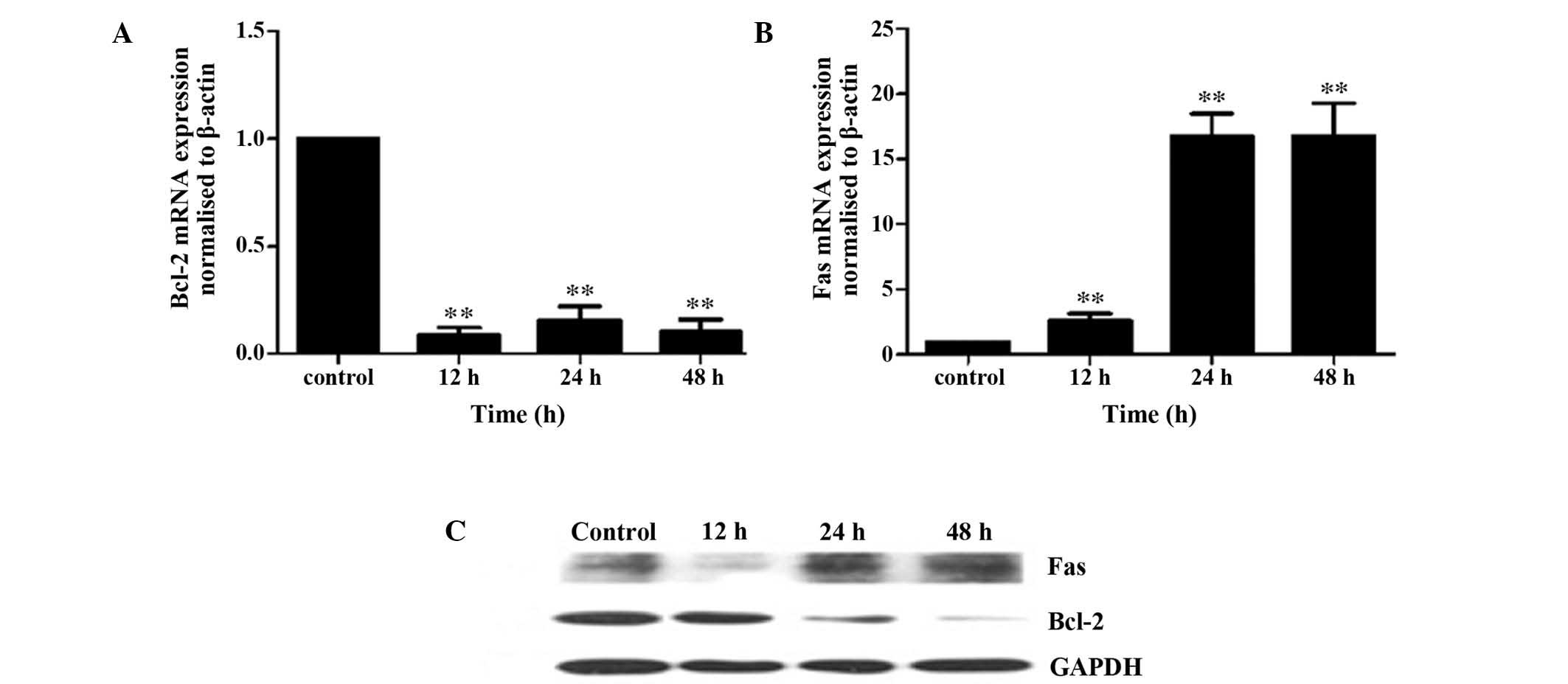
Mitochondrial and death receptor pathways
are involved in iodine-131-induced apoptosis
To assess the effect of iodine-131 treatment on the
expression of apoptosis-associated genes in HTori-3 cells, the
expression levels of Bcl-2 and Fas were determined using western
blot analysis and RT-qPCR. Fas was found to be transcriptionally
increased, while Bcl-2 mRNA expression decreased following
iodine-131 treatment (Figs. 4A and
B), determined by RT-qPCR. The increased Fas and decreased
Bcl-2 expression levels, following treatment with iodine-131, were
also observed by western blot analysis (Fig. 4C).
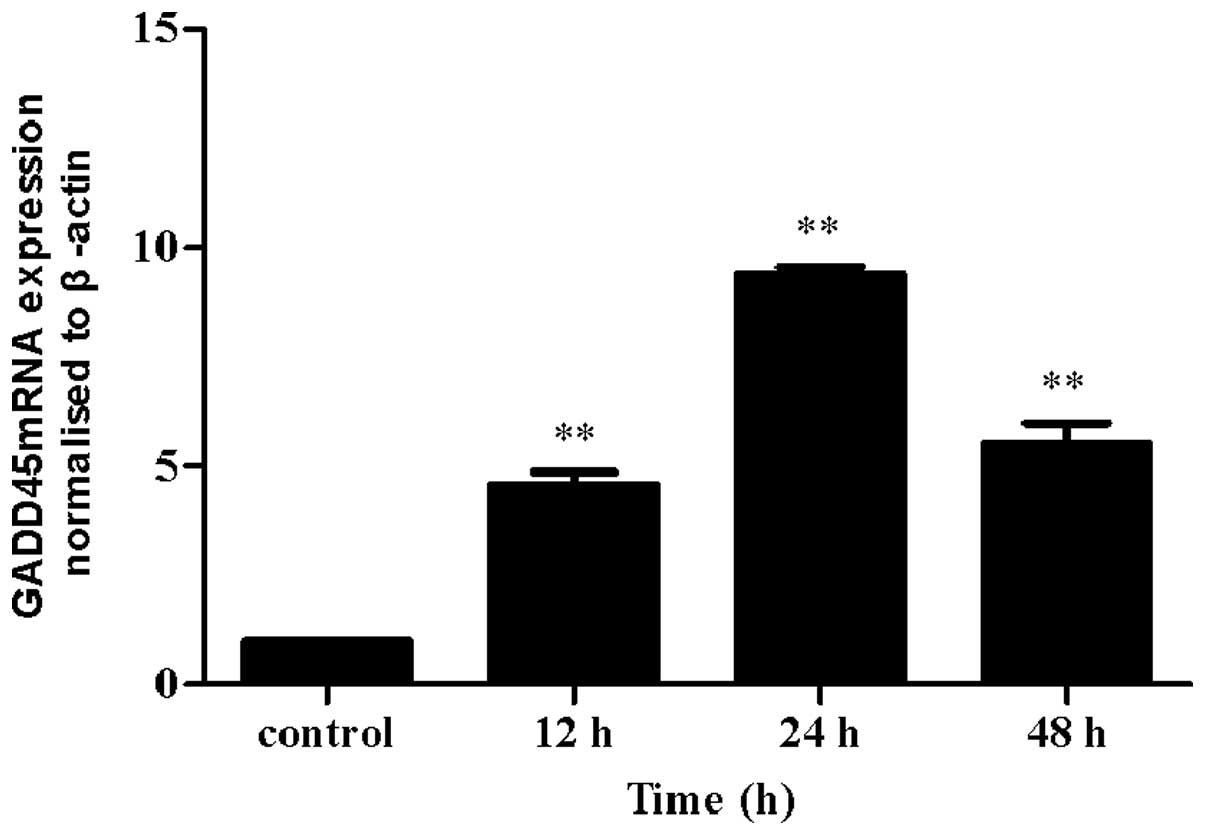
Iodine-131 induces GADD45 accumulation in
HTori-3 cells
To further examine whether the G2 phase arrest
induced by iodine-131 was correlated with GADD45 in the thyroid
cell line, the expression of GADD45 was evaluated by RT-qPCR prior
to and following iodine-131 exposure. A statistically significant
increase was observed in GADD45 transcription compared with the
control (Fig. 5).
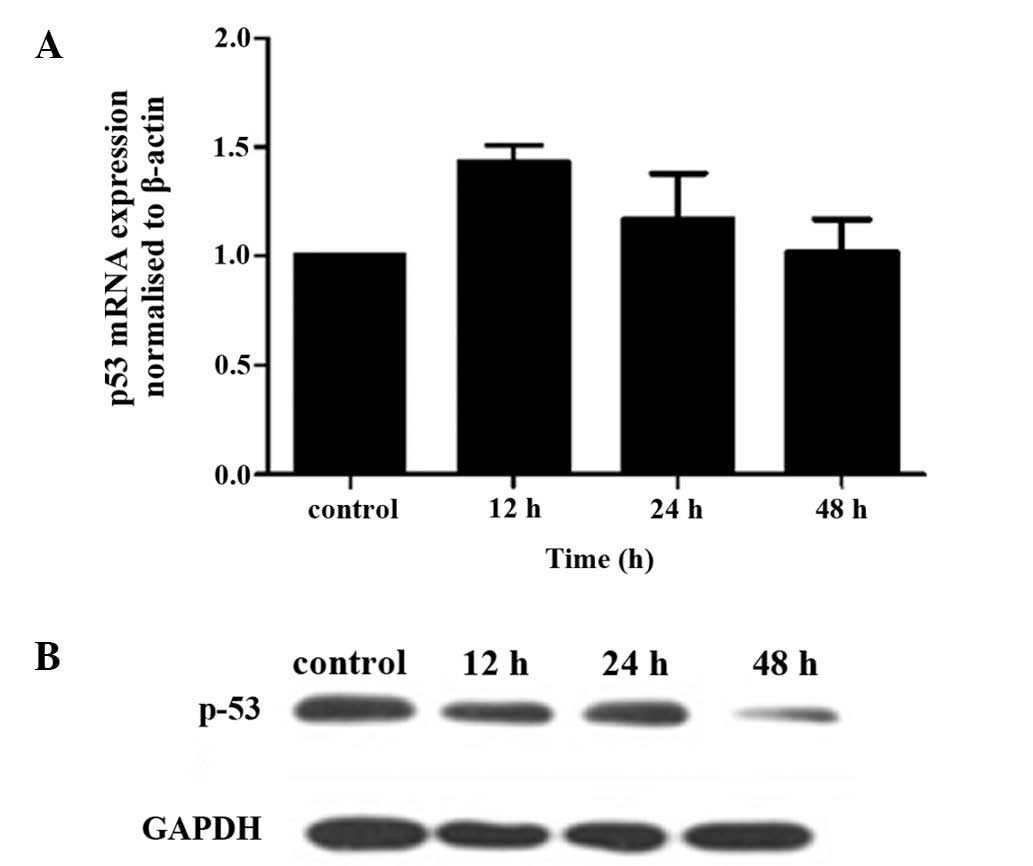
Iodine-131 results in HTori-3 cell growth
inhibition in a p53-independent manner
To investigate whether p53 was involved in the
iodine-131-induced inhibition of HTori-3 cell growth, mRNA and
protein expression levels of p53 were determined following cellular
stress induced by iodine-131. No changes were observed in p53 mRNA
expression following exposure to iodine-131 (Fig. 6A). In addition, iodine-131 did not
affect p53 protein expression, and even presented a reducing
tendency on p53 expression at 48 h after iodine-131 treatment
(Fig. 6B).
Discussion
The extent of DNA damage induced by ionizing
radiation depends on the type of radiation and the dose applied, as
well as other factors. Higher radiation linear energy transfer
results in greater complexity of the lesions, as well as more
challenging repair of the induced lesions (17,18).
The results of the present study revealed that the inhibitory
effect of iodine-131 on HTori-3 cell growth occurred via cell
apoptosis and G2/M phase arrest in a time- and dose-dependent
manner, indicating that the cytotoxic effect of iodine-131 is
associated with the irradiation time and dosage, as well as a
number of other factors, including cell type or oxygenation status,
which may require further study.
DNA damage induced by irradiation or drugs used for
cancer chemotherapy may lead to apoptotic cell death (19). Irradiated HTori-3 cells presented a
dose- and time-dependent increase in apoptosis. The Bcl-2 family is
divided into proapoptotic proteins, including Bax, and
antiapoptotic proteins, including Bcl-2 (10). Whether cell undergo apoptosis
depends on the ratio of Bcl-2 to Bax, to a certain extent (11). Fas is known to be critical in the
process of cell apoptosis induced by radiation damage and other DNA
damage (20,21). The intrinsic apoptotic pathway is
dominated by the Bcl-2 family of proteins (22), while the cell-surface receptor, Fas
(CD95/Apo-1) is a key component of the extrinsic death pathway
(20,21). The results of the present study
(Fig. 4) identified that treatment
of thyrocyte cells with iodine-131 resulted in decreased Bcl-2 and
significantly increased Fas expression levels, indicating that the
Bcl-2 and Fas expression levels are involved in signal transmission
in the process of cell apoptosis induced by β-ray radiation. In
addition, the apoptosis of HTori-3 cells, which was induced by
iodine-131, may occur through the death receptor pathway and the
mitochondrial pathway. Friesen et al (1) also demonstrated that β
radiation-induced and -activated apoptosis pathways in leukemia
cells by upregulating the expression levels of CD95 ligand and
receptor, as well as activating caspases. In human breast cancer
cells, apoptosis induced by strontium-89 is also regulated by the
decreased Bcl-2 and increased Fas expression levels (2).
G1/S and G2/M cell cycle checkpoints are essential
for the maintenance of genomic stability in response to DNA damage.
An evident effect was observed on cell cycle phase distribution in
the HTori-3 cell line due to the iodine-131 treatment, exhibiting a
positive correlation with the concentration and time of iodine-131
treatment. A number of previous studies have also reported that the
tumor cell cycle G2/M retardation was induced by γ-ray radiation
(23,24). This process was also identified in
the human breast carcinoma cell line, MCF-7, when induced by
89Sr and irradiated with β-rays (2). Eriksson et al (5) also demonstrated that HeLa Hep2 cells
exhibited a transient G2/M phase arrest following iodine-131
irradiation. GADD45, a DNA damage-inducible protein, has been
identified to be critical in the G2/M cell cycle checkpoint
(25). GADD45 mRNA expression was
found to be significantly increased following iodine-131 treatment
of HTori-3 cells (Fig. 5). These
results indicate that G2/M phase arrest mediated by GADD45 may be
one of the underlying mechanisms of the growth inhibitory effect of
iodine-131 on the HTori-3 cells.
The role and mechanism of the p53 tumor suppressor
protein on the cellular response to radiation-induced DNA damage is
well established. The p53 tumor suppressor gene is activated in the
cellular response to various cellular stresses, including
radiation-induced DNA damage, oncogene stimulation, hypoxia and
nucleotide depletion (26).
Activation of p53 results in cell growth arrest at G1 and/or G2
phase, DNA repair, senescence or apoptosis (8,9). In
the present study, to verify whether iodine-131 induced HTori-3
cell apoptosis in a p53-dependent pathway, the p53 mRNA and protein
expression levels were evaluated following iodine-131 treatment.
The results (Fig. 6) revealed that
the mRNA and protein expression levels of p53 were not altered
following iodine-131 exposure, indicating that apoptosis induced by
iodine-131 is independent of p53.
In conclusion, the current study demonstrated that
iodine-131 may induce apoptosis in HTori-3 cells by downregulating
the expression levels of the Bcl-2 antiapoptotic protein and
upregulating the mRNA and protein expression levels of Fas.
Furthermore, iodine-131 treatment may upregulate the expression of
GADD45 in the G2/M phase arrest in a p53-independent pathway.
Therefore, the results of the present study suggest that cell
apoptosis and cell cycle arrest in a p53-independent pathway may be
the underlying mechanism in iodine-131 therapy of hyperthyroidism.
Further investigation of the effects of β radiation at the cellular
level is required in order to enable its direct application in
nuclear medicine.
Acknowledgements
The authors would like to thank Professor Peng Hou
(Endocrinology Laboratory, The First Affiliated Hospital of Xi’an
Jiaotong University College of Medicine, Xi’an, China) for
providing useful advice. The project was supported by a grant from
the National Natural Science Foundation of China (no. 81172598).
The manuscript abstract was accepted as a meeting abstract by the
Society of Nuclear Medicine and Molecular Imaging 2014 Annual
Meeting (http://jnumedmtg.snmjournals.org/cgi/content/meeting_abstract/55/1_MeetingAbstracts/142)
(28).
References
|
1
|
Friesen C, Lubatschofski A, Kotzerke J,
Buchmann I, Reske SN and Debatin KM: Beta-irradiation used for
systemic radioimmunotherapy induces apoptosis and activates
apoptosis pathways in leukaemia cells. Eur J Nucl Med Mol Imaging.
30:1251–1261. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang C, Wang J, Jiang H, Zhu M, Chen B and
Bao W: In vitro study on apoptosis induced by strontium-89 in human
breast carcinoma cell line. J Biomed Biotechnol. 2011:5414872011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sawka AM, Thephamongkhol K, Brouwers M,
Thabane L, Browman G and Gerstein HC: Clinical review 170: A
systematic review and metaanalysis of the effectiveness of
radioactive iodine remnant ablation for well-differentiated thyroid
cancer. J Clin Endocrinol Metab. 89:3668–3676. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bogazzi F, Giovannetti C, Fessehatsion R,
et al: Impact of lithium on efficacy of radioactive iodine therapy
for Graves’ disease: a cohort study on cure rate, time to cure, and
frequency of increased serum thyroxine after antithyroid drug
withdrawal. J Clin Endocrinol Metab. 95:201–208. 2010. View Article : Google Scholar
|
|
5
|
Eriksson D, Blomberg J, Lindgren T,
Löfroth PO, et al: Iodine-131 induces mitotic catastrophes and
activates apoptotic pathways in HeLa Hep2 cells. Cancer Biother
Radiopharm. 23:541–549. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Marx K, Moka D, Schomäcker K, et al: Cell
death induced by 131I in a differentiated thyroid
carcinoma cell line in vitro: necrosis or apoptosis? Nucl Med
Commun. 27:353–358. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Reinhardt HC and Schumacher B: The p53
network: cellular and systemic DNA damage responses in aging and
cancer. Trends Genet. 28:128–136. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Feng Z and Levine AJ: The regulation of
energy metabolism and the IGF-1/mTOR pathways by the p53 protein.
Trends Cell Biol. 20:427–434. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jin S and Levine AJ: The p53 functional
circuit. J Cell Sci. 114:4139–4140. 2001.PubMed/NCBI
|
|
10
|
Brunelle JK and Letai A: Control of
mitochondrial apoptosis by the Bcl-2 family. J Cell Sci.
122:437–441. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shi L, Chen J, Yang J, Pan T, Zhang S and
Wang Z: MiR-21 protected human glioblastoma U87MG cells from
chemotherapeutic drug temozolomide induced apoptosis by decreasing
Bax/Bcl-2 ratio and caspase-3 activity. Brain Res. 1352:255–264.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Villa-Morales M, González-Gugel E,
Shahbazi MN, Santos J and Fernández-Piqueras J: Modulation of the
Fas-apoptosis-signalling pathway by functional polymorphisms at
Fas, FasL and Fadd and their implication in T-cell lymphoblastic
lymphoma susceptibility. Carcinogenesis. 31:2165–2171. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ji J, Tian Y, Ji S, Zhang L, Zhu Y and Lu
X: Ionizing radiation induces cell cycle G1 arrest and tumor
suppressor gene P16, P21, P27 expression in keloid fibroblasts. Int
J Radiat Oncol. 87(Suppl): S6292013. View Article : Google Scholar
|
|
14
|
Niehrs C and Schäfer A: Active DNA
demethylation by Gadd45 and DNA repair. Trends Cell Biol.
22:220–227. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pilli T, Prasad KV, Jayarama S, Pacini F
and Prabhakar BS: Potential utility and limitations of thyroid
cancer cell lines as models for studying thyroid cancer. Thyroid.
19:1333–1342. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Caudill CM, Zhu Z, Ciampi R, Stringer JR
and Nikiforov YE: Dose-dependent generation of RET/PTC in human
thyroid cells after in vitro exposure to γ-radiation: a model of
carcinogenic chromosomal rearrangement induced by ionizing
radiation. J Clin Endocrinol Metab. 90:2364–2369. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Moore S, Stanley FK and Goodarzi AA: The
repair of environmentally relevant DNA double strand breaks caused
by high linear energy transfer irradiation - no simple task. DNA
Repair (Amst). 17:64–73. 2014. View Article : Google Scholar
|
|
18
|
Reisz JA, Bansal N, Qian J, Zhao W and
Furdui CM: Effects of ionizing radiation on biological molecules -
mechanisms of damage and emerging methods of detection. Antioxid
Redox Signal. 10:260–292. 2014. View Article : Google Scholar
|
|
19
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Villa-Morales M and Fernández-Piqueras J:
Targeting the Fas/FasL signaling pathway in cancer therapy. Expert
Opin Ther Targets. 16:85–101. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Koster R, Timmer-Bosscha H, Bischoff R,
Gietema JA and de Jong S: Disruption of the MDM2-p53 interaction
strongly potentiates p53-dependent apoptosis in cisplatin-resistant
human testicular carcinoma cells via the Fas/FasL pathway. Cell
Death Dis. 2:e1482011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cory S and Adams JM: The Bcl2 family:
regulators of the cellular life-or-death switch. Nat Rev Cancer.
2:647–656. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sak A, Wurm R, Elo B, et al: Increased
radiation-induced apoptosis and altered cell cycle progression of
human lung cancer cell lines by antisense oligodeoxynucleotides
targeting p53 and p21 WAF1/CIP1. Cancer Gene Ther. 10:926–934.
2003. View Article : Google Scholar
|
|
24
|
Fernet M, Mégnin-Chanet F, Hall J and
Favaudon V: Control of the G2/M checkpoints after exposure to low
doses of ionising radiation: implications for
hyper-radiosensitivity. DNA Repair (Amst). 9:48–57. 2010.
View Article : Google Scholar
|
|
25
|
Zimmermann M, Arachchige-Don AS, Donaldson
MS, Dallapiazza RF, Cowan CE and Horne MC: Elevated cyclin G2
expression intersects with DNA damage checkpoint signaling and is
required for a potent G2/M checkpoint arrest response to
doxorubicin. J Biol Chem. 287:22838–22853. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fridman JS and Lowe SW: Control of
apoptosis by p53. Oncogene. 22:9030–9040. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang W, Gao R, Yu Y, Guo K, Liu Y and
Yang A: Radioactive iodine-131 induces human thyrocyte cell line
HTori 3 cell apoptosis and G2/M arrest in a p53-independent
pathway. J Nucl Med Meeting Abstracts. 55:1422014.
|