Introduction
The receptor for advanced glycation end products
(RAGE) is a multiligand receptor of the immunoglobulin (Ig)
superfamily of cell surface molecules, first introduced as a
critical factor in diabetes and other metabolic disorders
characterized by AGE accumulation. As well as binding with AGEs,
RAGE interacts with high-mobility group box protein 1, members of
the S100/calgranulin family, β2-integrin Mac, amyloid-β peptide and
β-sheet fibrils (1–3). A previous in vivo study
revealed that RAGE mRNA and protein expression significantly
increased in the coronary arteries of type II diabetic mice
compared with non-diabetic mice (4). AGE and RAGE expression were
upregulated at sites of endothelial denudation. This was
particularly prevalent in activated smooth muscle cells (SMCs) of
the expanding neointima in mice (5). SMC proliferation, migration and
neointimal expansion upon arterial injury were markedly suppressed
in homozygous RAGE null mice compared with wildtype litter mates
(5). Furthermore, RAGE
overexpression was associated with enhanced inflammatory reactions
and increased expression of cyclooxygenase-2, microsomal
prostaglandin synthase-1, matrix metalloproteinase-2 (MMP-2) and
MMP-9 in plaque macrophages and SMCs in diabetic patients (6).
The nuclear receptor peroxisome
proliferator-activated receptor γ (PPARγ) is a member of the
nuclear hormone receptor superfamily of ligand-activated
transcriptional factors. Previously, PPARγ activators were
demonstrated to exert anti-inflammatory, anti-oxidative and
anti-proliferative effects on vascular wall cells (7). A number of studies have revealed that
PPARγ agonists inhibit the deleterious effects of AGEs in cell
culture and animal models. Pretreatment of rat aortic SMCs with
PPARγ agonist rosiglitazone, significantly downregulated RAGE
expression and subsequently inhibited SMC proliferation in response
to treatment with RAGE agonists S100/calgranulin (7). Insulin inhibited AGE-induced SMC
proliferation by not only suppressing nuclear factor-κB (NF-κB)
activation, but also by increasing PPARγ expression (8). Furthermore, rosiglitazone was
reported to decrease RAGE expression and SMC proliferation
following carotid arterial injury in diabetic and non-diabetic rats
(7).
It remains to be elucidated if high glucose is able
to induce coronary artery SMC RAGE expression and the mechanism has
not been investigated. In addition, little is known with regard to
the effect of pioglitazone on high glucose-induced RAGE expression
in coronary artery vascular SMCs (VSMCs), which are the main cell
type in coronary atherosclerosis. In the present study, RAGE
expression in coronary artery SMCs was investigated following
treatment with varying concentrations of glucose. The effects of
the PPARγ agonist pioglitazone (PIO) on RAGE expression in VSMCs
and its underlying mechanism following treatment with high
concentrations of glucose was also investigated.
Materials and methods
Materials
Sprague-Dawley (SD) rats were purchased from Vital
River Laboratories Animals Technology Co., Ltd. (Beijing, China).
PIO was donated by Huadong Medicine Co., Ltd. (Hangzhou, China).
GW9662, diphenylene iodonium (DPI) and pyrrolidine dithiocarbamate
(PDTC) were purchased from Sigma (St. Louis, MO, USA). Dulbecco’s
modified Eagle medium (DMEM) and fetal bovine serum (FBS) were
purchased from Hyclone Laboratories, Inc. (Logan, UT, USA).
TRIzol® reagent and cell lysates were purchased from
Invitrogen Life Technologies (Shanghai, China). Anti-GAPDH was
purchased from ProMab Biotechnologies Inc. (Richmond, CA, USA)
Anti-RAGE was obtained from Abcam (Hong Kong, SAR, P.R. China).
Anti-NF-κB p65, anti-I-κBα, fluorescein isothiocyanate
(FITC)-labeled goat anti-rabbit IgG and the reactive oxygen species
(ROS) assay kit were purchased from the Beyotime Institute of
Biotechnology (Jiangsu, China).
Cell culture and treatment
Male SD rats (Vital River Laboratories Animals
Technology Co., Ltd.) weighing 250 g were housed in an
environmentally controlled room with a 12-h light/dark cycle and
administered standard rodent chow and tap water ad libitum.
The present study was approved by the ethics committee of Beijing
Friendship Hospital, Capital Medical University (Beijing, China).
Male SD rats at seven weeks old were anesthetized with
bentobarbital sodium (60 mg/kg). Rat coronary arteries were
dissected from the ventricle and the endothelium in the vessels was
denuded with air. Enzymatic isolation of VSMCs was performed
according to published methods (9). Coronary artery VSMCs were cultured in
DMEM supplemented with 15% fetal calf serum, 100 U/ml penicillin-G
and 100 mg/ml streptomycin. The morphology and growth
characteristics of the cells were typical of SMCs and were
identified as SMCs by positive α-smooth muscle actin staining.
Cultured cells were incubated in DMEM with 15% FBS, followed by
synchronization for 24 h with serum-deprived medium containing 5.5
mM D-glucose and 1% FBS.
To examine RAGE mRNA and protein expression in
coronary artery VSMCs incubated with different concentrations of
glucose, cells were cultured in 5.5, 12.0, 18.0 or 25.0 mmol/l
glucose-treated medium. Following incubation for 24 or 48 h,
western blot analysis and reverse-transcription quantitative
polymerase chain reaction (RT-qPCR) were performed. To determine
the effect of PIO, GW9662 (PPARγ antagonist), DPI (NADPH oxidase
inhibitor) and PDTC (NF-κB inhibitor) on high glucose-induced RAGE
expression in coronary artery VSMCs, cells were divided into the
following treatment groups: i) 5.5 mmol/l D-glucose (normal
glucose); ii) 25 mmol/l D-glucose (high glucose); iii) 25 mmol/l
D-glucose and 10 μmol/l PIO; iv) 25 mmol/l D-glucose, 10 μmol/l PIO
and 10 μmol/l GW9662; v) 25 mmol/l D-glucose and 10 μmol/l DPI and
vi) 25 mmol/l D-glucose and 40 μmol/l PDTC. Cells were incubated
for 48 h before western blotting and immunofluorescence to evaluate
RAGE protein expression and incubated for 24 h prior to RT-qPCR
analysis to evaluate RAGE mRNA expression.
The effects of PIO, GW9662, DPI and PDTC on high
glucose-induced NF-κB activation and ROS production in coronary
artery SMCs were evaluated. After 24 h preincubation in medium with
5.5 mmol/l D-glucose, coronary artery SMCs were exposed to the
following experimental conditions for 24 h: i) 5.5 mmol/l D-glucose
(normal glucose); ii) 25 mM D-glucose (high glucose); iii) 25
mmol/l D-glucose and 10 μmol/l PIO; iv) 25 mmol/l D-glucose, 10
μmol/l PIO and 10 μmol/l GW9662; v) 25 mmol/l D-glucose and 10
μmol/l DPI; vi) 25 mM D-glucose and 40 μmol/l PDTC. Following
incubation, immunofluorescence was used to analyze the nuclear
translocation of NF-κB and ROS levels were assessed using the
CM-H2DCFDA fluoroprobe.
RT-qPCR
Total RNA samples were extracted from rat coronary
artery SMCs with TRIzol® reagent (Invitrogen Life
Technologies). Total RNA was reverse-transcribed with the cDNA
synthesis kit according to the manufacturer’s instructions
(Promega, Sunnyvale, CA, USA). qPCR was performed with 5 μl
SYBR® Green 2× Realtime PCR master mix, 10 μmol/l
forward primer 0.2 μl, 10 μmol/l reverse primer 0.2 μl, 1 μl cDNA
and 3.5 μl double-distilled H20, for a total reaction
volume of 10 μl. Following initial denaturation at 95°C for 3 min,
PCR was performed for a total of 40 cycles, each at 95°C for 10
sec, 60°C for 10 sec and 72°C for 15 sec. The primers used were as
follows: Rat-RAGE, forward 5′-AGGAGGAGACCAGGAGGCACCC-3′ and reverse
5′-CTCCCTGACTCGGGGCTGGATG-3′; rat GAPDH, forward
5′-CAAGATTGTCAGCAATGCATCC-3′ and reverse
5′-ATCACGCCACAGCTTTCCAGAG-3′. The starting copy number of the
unknown samples was determined relative to the known copy number of
the calibrator sample using the following formula: ΔΔCt
= [Ct target gene (unknown sample)-Ct GAPDH gene (unknown
sample)]-[Ct target gene (calibrator sample)-Ct GAPDH gene
(calibrator sample)]. In this case, the target gene was RAGE. The
relative gene copy number was calculated by ΔΔCt. qPCR
data were normalized to an internal control (GAPDH) and were
presented as the mean ± standard deviation for three independent
experiments performed in triplicate.
Western blot analysis
Cells were lysed in 1 ml cell lysate (Total Protein
Extraction kit; ProMab) for 30 min at 4°C. The cell lysates were
centrifuged at 12556 × g for 30 min at 4°C to remove insoluble
material. The resulting supernatant was frozen at −80°C for later
analyses by SDS-PAGE and immunoblotting. A total amount of 25 μl
cell lysate were resolved on 12% SDS-PAGE gels (Sigma), followed by
electrophoretic transfer onto polyvinylidene fluoride membranes
(Pierce Biotechnology, Inc., Rockford, IL, USA). The membranes were
blocked with 5% non-fat milk blocking buffer and then incubated
overnight at 4°C with primary antibody (1:500 dilution; Abcam).
Following extensive washing with Tris-buffered saline (TBS)
containing 0.5% Tween 20 (Beyotime Institute of Biotechnology),
membranes were incubated with horseradish peroxidase-labeled
secondary antibody (1:4,000 dilution; Thermo Pierce, Rockford, IL,
USA) at room temperature for 1 h, followed by additional washes
with TBS containing 0.5% Tween 20. The blots were developed using a
chemiluminescence kit. Densitometric analyses of autoradiographic
bands were normalized to GAPDH expression, taking into account the
size and area of the bands (Scion Image software (Scion Corp.,
Frederick, MD, USA).
Immunofluorescence microscopy
Coronary artery SMCs grown on glass coverslips were
washed in phosphate-buffered saline (PBS; Hyclone Laboratories,
Inc.) and fixed in 4% paraformaldehyde (10 min at room temperature;
Beyotime Institute of Biotechnology). Non-specific binding sites
were blocked in 1% BSA (Hyclone Laboratories, Inc.) for 2 h. Cells
were then incubated with a rabbit polyclonal anti-RAGE antibody
(1:100 in blocking buffer) or a rabbit polyclonal anti-NF-κB p65
antibody (1:100 in blocking buffer). Following washing, cells were
exposed to a goat anti-rabbit IgG-FITC (1:200) for 1 h at room
temperature. Cells were mounted in 80% glycerol (Beyotime Institute
of Biotechnology) and observed under a confocal microscope (LSM510;
Zeiss, Oberkochen, Germany).
Measurement of ROS levels
Coronary artery SMCs were stimulated, harvested by
trypsinization, resuspended in PBS at a concentration of
106 cells/ml and loaded with 10 mol/l CM-H2DCFDA
(Beyotime Institute of Biotechnology). Dichlorofluorescein (DCF;
Beyotime Institute of Biotechnology) fluorescence was monitored by
analyzing 5,000 cells in a flow cytometer (Nikon, Tokyo,
Japan).
Statistical analysis
Data are expressed as the mean ± standard deviation.
Results were analyzed using one-way analysis of variance for
multiple comparisons, followed by the Fisher’s least significant
difference test. Statistical analyses were performed using SPSS
16.0 software (SPSS, Inc., Chicago, IL, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
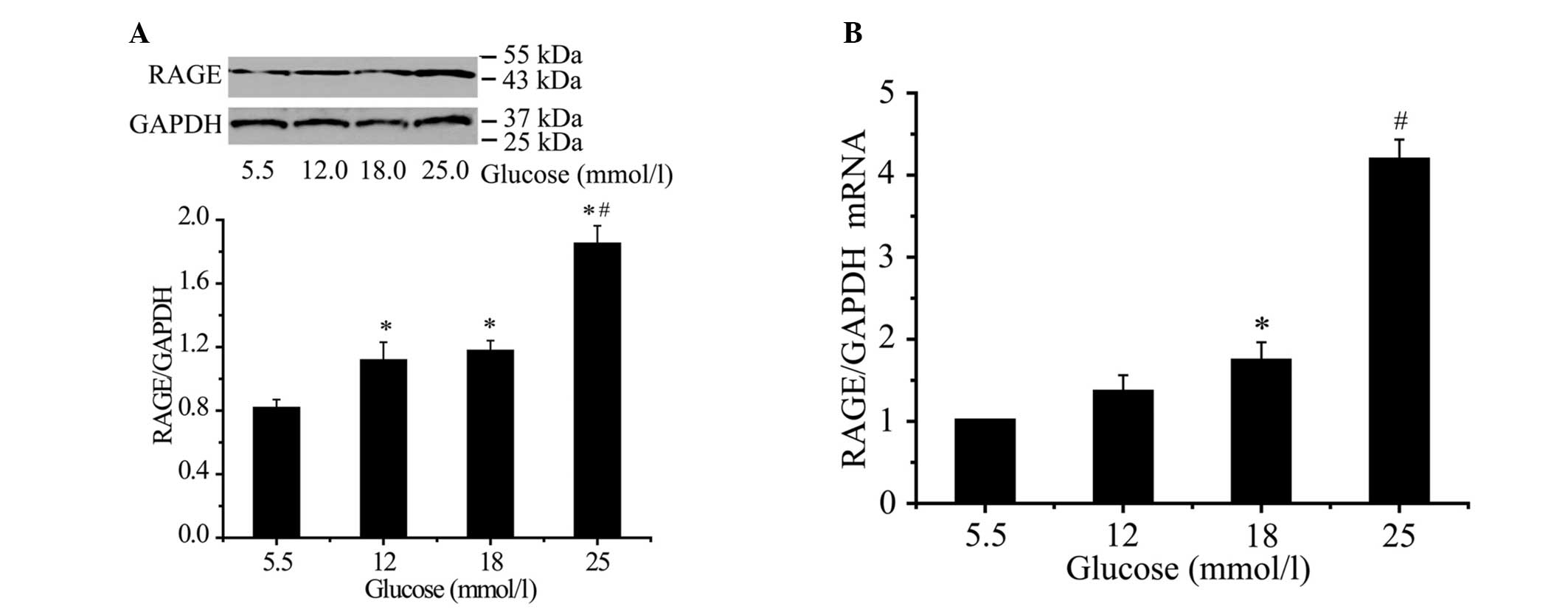
Effect of glucose on RAGE protein and
mRNA expression in coronary artery SMCs
RAGE protein expression increased by 37.0, 44.4 and
127% at concentrations of 12 mmol/l (P<0.05), 18 mmol/l
(P<0.05) and 25 mmol/l (P<0.01), respectively, following a 48
h incubation (Fig. 1A). RAGE mRNA
expression increased by 35.1, 73.1 and 318.0% at concentrations of
12 mmol/l (P<0.05), 18 mmol/l (P<0.05) and 25 mmol/l
(P<0.01), respectively, after a 24-h incubation (Fig. 1A). The effect of glucose on RAGE
mRNA and protein expression was concentration dependent. Maximal
glucose treatment was reached at 25 mmol/l (P<0.01; Fig. 1B).
PIO, DPI and PDTC decreases high
glucose-induced RAGE expression of coronary artery VSMCs
It was identified that 10 μmol/l PIO (PPARγ
agonist), 10 μmol/l DPI (NADPH oxidase inhibitor) and 40 μmol/l
PDTC (NF-κB inhibitor) significantly inhibited high
glucose-stimulated RAGE expression. RAGE mRNA expression declined
by 65.2, 70.7 and 56.1% following pretreatment with PIO, DPI or
PDTC respectively (Fig. 2A). RAGE
protein expression decreased by 52.8, 58.9 and 50.1% following
pretreatment with PIO, DPI, or PDTC, respectively (Fig. 2B). These findings suggest that high
glucose-induced RAGE expression is dependent upon activation of
NADPH oxidase, ROS generation and NF-κB activation. PIO may mimic
the effect of DPI or PDTC, significantly downregulating high
glucose-stimulated RAGE expression. Pretreatment of PIO with GW9662
induced the lower expression of RAGE compared with treatment with
PIO only, which indicates that PIO inhibits RAGE expression through
interaction with PPARγ. The immunofluorescence results were in
accordance with our previous assessment of RAGE protein expression
by western blot analysis (Fig.
2C).
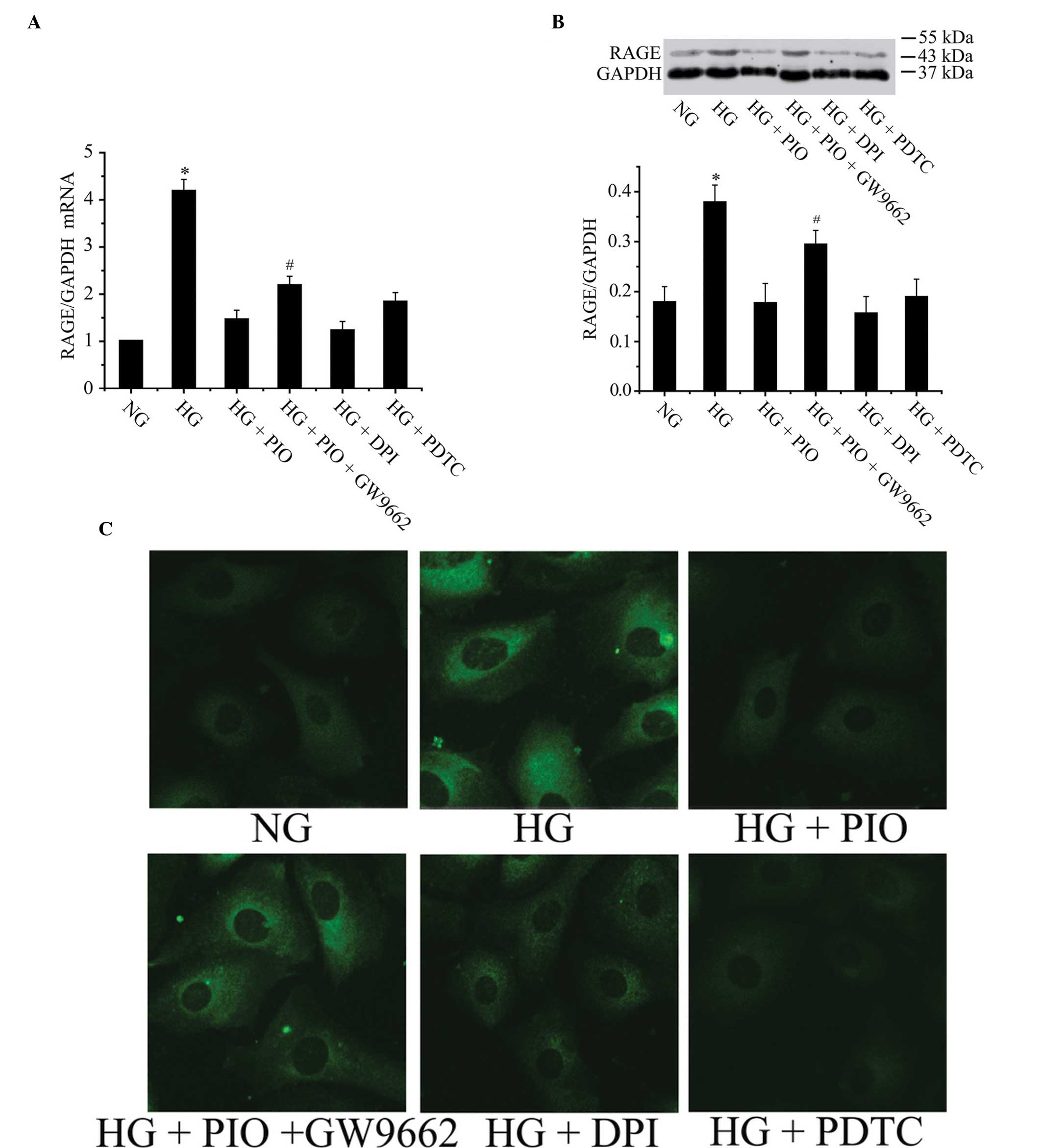 | Figure 2PIO, DPI and PDTC decrease HG-induced
RAGE expression in coronary vascular smooth muscle cells. (A) RAGE
mRNA declined significantly following pretreatment with PIO, DPI or
PDTC, respectively. Pretreatment of PIO plus GW9662 induced
increased RAGE mRNA expression compared with treatment only by PIO.
*P<0.05 vs. the NG, HG+PIO, HG+DPI or HG+PDTC groups;
#P<0.05 vs. the HG+PIO group. (B) PIO, DPI and PDTC
significantly inhibited HG-stimulated RAGE expression. Pretreatment
of PIO plus GW9662 induced increased expression level of RAGE
compared with treatment of only PIO. *P<0.05 vs.
other groups; #P<0.05 vs. HG+PIO group. (C) RAGE
associated immunofluorescence result was in agreement with previous
observations of RAGE expression as assessed by western blot
analysis (magnification, ×400). NG, normal glucose; HG, high
glucose; PIO, pioglitazone; PDTC, pyrrolidine dithiocarbamate; DPI,
diphenyleneiodonium; RAGE, receptor for advanced glycation end
products; NG, normal glucose; HG, high glucose. |
PIO, DPI and PDTC decreases high
D-glucose-induced NF-κB activation of coronary artery SMCs
The mechanism by which PIO inhibited high
glucose-induced RAGE expression in coronary artery VSMCs was
further examined. It has been observed that the RAGE promoter
possesses NF-κB binding sites. It was therefore evaluated whether
PIO functioned by affecting the NF-κB pathway. High glucose
treatment significantly decreased cytoplasmic IκBα protein levels.
When PIO was included in the culture medium, this effect was
inhibited. Furthermore, addition of GW9662 eradicated the
inhibitory effect of PIO (Fig.
3A). Immunofluorescence was used to detect the nuclear
translocation of NF-κB in coronary artery VSMCs cells in response
to high glucose (Fig. 3B). NF-κB
primarily existed in the cytoplasm of control cells, but
translocated from the cytoplasm to the nucleus following treatment
with 25 mmol/l glucose. However, PIO inhibited high glucose-induced
NF-κB translocation and this inhibition was eradicated with GW9662
(Fig. 3B). DPI and PDTC were also
able to inhibit high glucose-induced NF-κB translocation. Taken
together, the present findings demonstrate that PIO may inhibit
NF-κB signaling through the PPARγ receptor in response to high
glucose levels, which partially explains the inhibitory effect of
PIO on high glucose-induced RAGE expression in coronary artery
VSMCs.
 | Figure 3Peroxisome proliferator-activated
receptor γ agonist PIO, nicotinamide adenine dinucleotide phosphate
oxidase inhibitor DPI and NF-κB inhibitor PDTC decrease high
D-glucose-induced NF-κB activation of coronary artery smooth muscle
cells. (A) HG treatment significantly decreased cytoplasmic I-κBα
protein levels. PIO, DPI or PDTC were able to inhibit this effect.
Furthermore, addition of GW9662 eliminated the inhibitory effect of
PIO. *P<0.05 vs. the other groups;
#P<0.05 vs. the HG+PIO group. (B) PIO inhibited
HG-induced NF-κB translocation and the inhibition was eradicated by
GW9662. DPI and PDTC may also inhibit HG-induced NF-κB
translocation (magnification, ×400). NG, normal glucose; HG, high
glucose; PIO, pioglitazone; PDTC, pyrrolidine dithiocarbamate; DPI,
diphenyleneiodonium; RAGE, receptor for advanced glycation end
products; NG, normal glucose; HG, high glucose; NF-κB, nuclear
factor-κB; I-κBα, NF-κB inhibitor α. |
PIO and DPI decrease high
D-glucose-induced ROS production
To investigate whether PIO may decrease ROS levels,
coronary artery SMCs were treated with high glucose (25 mmol/l) for
24 h and ROS levels were measured by DCF fluorescence in a flow
cytometer. Significantly increased levels of ROS compared with
control cells were observed following treatment with high glucose.
Pretreatment with DPI or PIO significantly decreased high
glucose-induced ROS elevation, as measured by DCF fluorescence
(Fig. 4).
Discussion
A large body of experimental evidence supports the
integral contribution of RAGE activation to all major stages
associated with the development and progression of atherosclerosis
in diabetes. RAGE activation was able to elicit ROS generation and
subsequently induce SMC proliferation (10,11).
The source of RAGE activation mediated ROS production is supposedly
NADPH oxidase and in cultured SMCs, various downstream signaling
pathways, including NF-κB, are activated by stimulation of RAGE
(12). AGEs stimulate collagen
synthesis activity and induce cell-associated fibronectin
production and TGF-β expression in cultured SMCs through
interaction with RAGE (13). The
AGE-RAGE interaction stimulates expression of typical bone
proteins, including alkaline phosphatase, osteopontin and
osetocalcin in cultured SMCs and therefore increases calcification
in the arteries (14–16).
It has been demonstrated that the expression of RAGE
in coronary arterioles was markedly increased in diabetic vs.
control mice, but the underlying mechanism remains to be
elucidated. One of the major findings of the present study is that
glucose was able to directly induce RAGE protein and mRNA
expression of coronary artery SMCs in a concentration-dependent
manner, which is one of the mechanisms underlying elevated RAGE
expression in the coronary arteries of diabetic mice. Former
studies revealed that in diabetic RAGE−/−/apolipoprotein
E (ApoE)−/− double knockout mice, the absence of RAGE
was associated with the significant attenuation of atherosclerotic
plaque accumulation. Overexpression of RAGE induces the expression
of a number of proinflammatory genes (MCP-1, IL-6 and ICAM-1) in
VSMCs (17). Overexpression of
RAGE in coronary artery VSMCs induced by high glucose may be
critical in accelerating inflammation and atherosclerosis of
coronary arteries.
In diabetic RAGE−/−/ApoE−/−
double knockout mice, expression of the aortic NF-κB p65 subunit,
inflammatory cytokines, adhesion molecules, including VCAM-1, MCP-1
and certain NADPH oxidase subunits, including gp91phox, p47phox and
rac-1 were decreased significantly when compared with diabetic
ApoE−/− mice (18–20).
These studies not only illustrated the importance of RAGE in
atherosclerosis progression in diabetic mice, but also indicated
that RAGE activation is closely associated with the NF-κB pathway
and ROS originating from NAPDH oxidase. The association between
increased expression of RAGE and elevated ROS production as well as
NF-κB activation remains to be investigated further.
In the present study, the mechanism underlying high
glucose-induced RAGE expression was investigated. In accordance
with former studies, high glucose was able to induce ROS production
following treatment for 12 h. Following inhibition of NADPH oxidase
by DPI, high glucose-induced ROS production and RAGE expression in
coronary artery SMCs significantly decreased compared with the high
glucose treatment group. Therefore, it is hypothesized that high
glucose elicits the generation of ROS and subsequently induces RAGE
expression in coronary artery SMCs. Previous studies have
demonstrated that the RAGE promoter possesses NF-κB binding sites
(21). In the present study, NF-κB
activation was significantly inhibited by PDTC and DPI; inhibition
of NF-κB activation by PDTC was able to downregulate high
glucose-induced RAGE expression. Thus, it is hypothesized that high
glucose upregulates RAGE expression by stimulating ROS production
and activating the NF-κB signaling pathway.
PIO is an anti-diabetic insulin-sensitizing agent
that improves insulin action in a variety of animal models of
insulin resistance and diabetes. It is considered to exert such
effects by acting as a selective ligand of PPARγ receptors. In
addition, previous studies have demonstrated that PIO not only
ameliorates insulin sensitivity, but also has anti-inflammatory,
antioxidative and antiproliferative effects on vascular wall cells.
It has been demonstrated that ligands of PPARγ receptors, including
PIO inhibit VSMC ROS production and inflammation, which are
important in the process of atherosclerosis formation in diabetes.
Previous studies have revealed that PIO inhibits AGE-induced VSMC
proliferation via increasing PPARγ expression and inhibiting the
ROS/ERK1/2 signaling pathway (22). In the current study, it was
observed that PIO decreased high glucose-induced upregulation of
RAGE expression in coronary artery VSMCs, whereas pretreatment of
SMCs with PIO and GW9662 did not downregulate RAGE expression.
These findings raised the possibility that PIO may decrease RAGE
expression in coronary artery SMCs through a PPARγ-dependent
mechanism. The mechanism of RAGE downregulation by a PPARγ agonist
was also investigated. In cultured coronary artery SMCs, it was
observed that high glucose potently induced intracellular ROS
production. DPI pretreatment significantly attenuated RAGE
expression induced by high glucose by scavenging ROS production.
Additionally, PIO was able to mimic the effects of DPI in coronary
artery SMCs. These results suggest that alleviating oxidative
stress using PIO is able to inhibit RAGE expression in coronary
artery SMCs.
In the present study, it was observed that PDTC, DPI
and PIO inhibited NF-κB activation in high glucose-treated VSMCs.
Furthermore, pharmacological inhibition of NF-κB activation by PDTC
or treatment with PIO significantly downregulated high
glucose-induced RAGE expression in coronary artery SMCs. Taken
together, these results suggest that PIO inhibits high
glucose-induced RAGE expression through inhibiting ROS-mediated
NF-κB activation. In conclusion, the present study demonstrated
that high glucose may directly induce RAGE expression through
stimulating ROS-mediated NF-κB activation, which may be inhibited
by PIO. These data provide novel insights into the molecular
pathways underlying increased RAGE expression in the coronary
artery SMCs stimulated by high glucose, and reveal novel mechanisms
that contribute to the beneficial effects of PIO in coronary
atherosclerosis in diabetes.
Acknowledgements
This study was supported by the Beijing Natural
Science Foundation (grant no. 7122053). PIO was donated by Huadong
Medicine Company (Hangzhou, China).
References
|
1
|
Hofmann MA, Drury S, Fu C, et al: RAGE
mediates a novel proinflammatory axis: A central cell surface
receptor for S100/calgranulin polypeptides. Cell. 97:889–901. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bierhaus A and Nawroth PP: Multiple levels
of regulation determine the role of the receptor for AGE (RAGE) as
common soil in inflammation, immuneresponses and diabetes mellitus
and its complications. Diabetologia. 52:2251–2263. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Leclerc E, Fritz G, Vetter SW and Heizmann
CW: Binding of S100 proteins to RAGE: An update. Biochim Biophys
Acta. 1793:993–1007. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gao X, Zhang HR, Schmidt AM and Zhang C:
AGE/RAGE produces endothelial dysfunction in coronary arterioles in
Type 2 diabetic mice. Am J Physiol Heart Circ Physiol.
295:H491–H498. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sakaguchi T, Yan SF, Yan SD, et al:
Central role of rage dependent neointimal expansion in arterial
restenosis. J Clin Invest. 111:959–972. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cipollone F, Iezzi A, Fazia M, et al: The
receptor rage as a progression factor amplifying
arachidonate-dependent inflammatory and proteolytic response in
human atherosclerotic plaques: Role of glycemic control.
Circulation. 108:1070–1077. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang K, Zhou Z, Zhang M, et al: Peroxisome
proliferator activated receptor gamma down-regulates receptor for
advanced glycation end products and inhibits smooth muscle cell
proliferation in a diabetic and nondiabetic rat carotid artery
injury model. J Pharmacol Exp Ther. 317:37–43. 2006. View Article : Google Scholar
|
|
8
|
De Oliveira C, Colette C, Monnier L,
Descomps B and Pares-Herbute N: Insulin alters nuclear
factor-lambdab and peroxisome proliferator-activated receptor-gamma
protein expression induced by glycated bovine serum albumin in
vascular smooth-muscle cells. J Lab Clin Med. 145:1440–1450. 2005.
View Article : Google Scholar
|
|
9
|
Li H, Chai Q, Gutterman DD and Liu Y:
Elevated glucose impairs cAMP mediated dilation by reducing Kv
channel activity in rat small coronary smooth muscle. Am J Physiol
Heart Circ Physiol. 285:H1213–H1219. 2003.PubMed/NCBI
|
|
10
|
Yoon SJ, Yoon YW, Lee BK, et al: Potential
role of hmg coareductase inhibitor on oxidative stress induced by
advanced glycation endproducts in vascular smooth muscle cells of
diabetic vasculopathy. Exp Mol Med. 41:802–811. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yoon YW, Kang TS, Lee BK, et al:
Pathobiological role of advanced glycation endproducts via
mitogen-activated protein kinase dependent pathway in the diabetic
vasculopathy. Exp Mol Med. 40:398–406. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
San Martin A, Foncea R, Laurindo FR, et
al: Nox1-based nadph oxidase-derived superoxide is required for
vsmc activation by advanced glycation end-products. Free Radic Biol
Med. 42:1671–1679. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sakata N, Meng J and Takebayashi S:
Effects of advanced glycation end products on the proliferation and
fibronectin production of smooth muscle cells. J Atheroscler
Thromb. 7:169–176. 2000. View Article : Google Scholar
|
|
14
|
Ren X, Shao H, Wei Q, Sun Z and Liu N:
Advanced glycation end products enhance calcification in vascular
smooth muscle cells. J Int Med Res. 37:847–854. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tanikawa T, Okada Y, Tanikawa R and Tanaka
Y: Advanced glycation end products induce calcification of vascular
smooth muscle cells through rage/p38 mapk. J Vasc Res. 46:572–580.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wei Q, Ren X, Jiang Y, Jin H, Liu N and Li
J: Advanced glycation end products accelerate rat vascular
calcification through RAGE/oxidative stress. BMC Cardiovasc Disord.
13:132013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hayakawa E, Yoshimoto T, Sekizawa N,
Sugiyama T and Hirata Y: Overexpression of receptor for advanced
glycation end products induced monocyte chemoattractant protein-1
expression in rat vasculat smooth muscle cell line. J Atheroscler
Thromb. 19:13–22. 2012. View
Article : Google Scholar
|
|
18
|
Soro-Paavonen A, Watson AM, Li J, Paavonen
K, et al: Receptor for advanced glycation end products (RAGE)
deficiency attenuates the development of atherosclerosis in
diabetes. Diabetes. 57:2461–2469. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bu DX, Rai V, Shen X, et al: Activation of
the ROCK1 branch of the transforming growth factor-beta pathway
contributes to RAGE-dependent acceleration of atherosclerosis in
diabetic ApoE-null mice. Circ Res. 106:1040–1051. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ueno H, Koyama H, Shoji T, et al: Receptor
for advanced glycation end-products (RAGE) regulation of adiposity
and adiponectin is associated with atherogenesis in apoE-deficient
mouse. Atherosclerosis. 211:431–436. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li J and Schmidt AM: Characterization and
functional analysis of the promoter of RAGE, the receptor for
advanced glycation end products. J Biol Chem. 272:16498–16506.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yuan X1, Zhang Z, Gong K, Zhao P, Qin J
and Liu N: Inhibition of reactive oxygen species/extracellular
signal-regulated kinases pathway by pioglitazone attenuates
advanced glycation end products-induced proliferation of vascular
smooth muscle cells in rats. Biol Pharm Bull. 34:618–623. 2011.
View Article : Google Scholar : PubMed/NCBI
|