1. Introduction
In the adult skeletal system, bone is constantly
renewed by the removal of old bone and the production of new bone
tissue, a process mediated by osteoclasts and osteoblasts (1). This process requires the removal of
trenches and tunnels of bone from the surfaces of trabeculae and
cortical bone by osteoclasts (2).
Osteoblasts subsequently fill these trenches by generating novel
bone matrices within them (3).
Regulation of the proliferation and activation of osteoclasts and
osteoblasts results in modulation of the levels of bone resorption
and formation (1). Various
diseases, including osteoporosis, Paget’s disease and rheumatoid
arthritis (RA), may disturb the normal bone balance (4). Osteoclasts have a significant role in
diseases that induce bone loss. The discovery of the receptor
activator of nuclear factor-κB ligand (RANKL)/RANK/osteoprotegerin
(OPG) signaling pathway in the late 1990s was a breakthrough in the
elucidation of the regulatory mechanisms underlying
osteoclastogenesis and bone resorption (5,6). The
binding of RANKL to its receptor, RANK, results in the fusion,
differentiation, activation and survival of osteoclasts (6).
OPG functions as a decoy receptor for RANKL, which
prevents RANKL binding to RANK. OPG is therefore considered a
protective factor against bone loss (7). During the last few decades, studies
investigating the RANKL/RANK/OPG system have confirmed that the
RANKL/RANK/OPG system is a master regulator of bone resorption
(4). The RANKL/RANK/OPG system
also has functions in other tissues, including as an
immunomodulator and thermoregulator (8,9).
2. Identification of RANKL/RANK/OPG
Prior to the 1990s it was known that osteoclasts
originated from mononuclear precursors in the myeloid lineage of
hematopoietic cells, which also produce macrophages. Osteoclast
progenitors (OCP) are able to differentiate into osteoclasts
following macrophage-colony stimulating factor (M-CSF) expression
by osteoblasts/stromal cells. The phenomenon that mice developed
osteopetrosis due to a lack of osteoclasts was identified in M-CSF
knockout mice, which indicated that M-CSF was essential to
osteoclastogenesis (10). However,
M-CSF expressed by osteoblasts and stromal cells is unable to
generate mature osteoclasts from osteoclast progenitor cells alone,
indicating that additional factors are also essential for this
process (6,11). These additional factors, expressed
by osteoblasts, stromal cells or other cell types, remained elusive
prior to the discovery of the RANKL/RANK/OPG system. The system was
discovered independently by various groups using differing
approaches (6,11,12).
OPG
The discovery of OPG was made independently by two
groups. In 1997, Simonet et al (7) in the USA identified a novel molecule
when they analyzed complementary DNA from the mouse intestine. The
group subsequently revealed that transgenic mice overexpressing the
OPG gene developed osteopetrosis due to a decrease in the number of
osteoclasts, which confirmed that OPG had a significant role in
osteclastogenesis.
Almost simultaneously, Rodan and Martin (13) in Japan reported the same molecule
independently. However, they had identified the molecule via an
alternative approach. In the early 1980s, Rodan and Martin
(13) hypothesized that
osteoblasts regulated osteoclast formation, and that factors
expressed by osteoblasts within bone are produced in response to
known stimulators of bone resorption. Subsequent studies supported
this hypothesis; however, the specific factors regulating
osteoclastogenesis remained elusive. Rodan and Martin continued
their search for these factors until they isolated a molecule that
inhibited osteoclastogenesis, which was revealed to be identical to
the molecule identified by Simonet et al (7). The discovery of OPG facilitated
further studies into the mechanisms underlying
osteoclastogenesis.
OPG is a member of tumor necrosis factor (TNF)
receptor superfamily (TNFRS), also known as TNFRS member 11B
(TNFRS11B) or osteoclastogenesis inhibitory factor (OCIF) and has a
similar domain to CD40; therefore, it is able to bind to CD40
ligand. The original OPG comprises a 401 amino acid peptide with a
21-amino acid propeptide that is cleaved, resulting in a mature
protein of 380 amino acids (7,14).
In contrast to other members of the TNFRS, OPG lacks transmembrane
and cytoplasmic domains and is highly expressed in soluble form in
order to function as a decoy receptor for RANKL (7). OPG is expressed in numerous types of
tissue, including the adult lung, heart, kidney, liver, thymus,
lymphnodes, bone marrow, osteoblasts, vascular smooth muscle cells,
B-lymphocytes and articular chondrocytes (15,16).
However, the function of OPG in a number of these tissues has
remained elusive.
Transgenic mice overexpressing OPG exhibited
osteopetrosis, in contrast to OPG-deficient mice, which developed
osteoporosis (16). The
osteoprotective role of OPG was further confirmed by the
identification of a 100-kb homozygous deletion of OPG in juvenile
Paget’s disease and an inactivating deletion in exon three of OPG
in idiopathic hyperphosphatasia (17).
RANKL
OPG was immediately used as a probe to identify its
ligand by the two groups who first reported the existence of OPG.
They named the ligand OPG ligand (OPGL) and osteoclast
differentiation factor (ODF), respectively (6,11).
At present, OPGL/ODF is more commonly known as RANKL.
RANKL is a member of the TNF family and is highly
conserved between species (7).
Human RANKL is closely associated with TNF-related
apoptosis-inducing ligand and Fas ligand, sharing ~34% and ~28%
sequence homology, respectively (6).
The RANKL gene is located on human chromosome 13q14
and spans ~36 kb of genomic DNA, comprising six exons (8).
Human RANKL is a 317-amino acid peptide, which forms
a 45 kDa membrane-associated protein. The soluble protein form of
RANKL has a molecular weight of 31 kDa and is cleaved by matrix
metalloproteinases (MMP)3 or 7 or a disintegrin and
metallopeptidase, rendering it soluble but less active (18).
RANKL is expressed in numerous tissues, including
bone and bone marrow, as well as lymphoid tissues (lymph nodes,
thymus, spleen, fetal liver and Peyer’s patches), mammary ligands
and the brain, which suggested potential functions beyond those in
bone (8,19). RANKL is a key regulator of
osteoclastogenesis and mice lacking RANKL developed osteopetrosis
due to osteoclast deficiency (20). The presence of M-CSF and RANKL was
demonstrated to be necessary and sufficient for the complete
differentiation of osteoclast precursor cells into mature
osteoclasts (6,11). The expression of RANKL by synovial
cells and activated T cells in RA contributes to bone loss and
inflammation of the joints, characteristic of the disease (21). Therefore, RANKL presents a
potential therapeutic target against bone destruction in RA.
RANK
Following the discovery of OPG and RANKL, the
receptor of RANKL required identification. However, the receptor
for RANKL had previously been identified as RANK (8). Human RANK is an amino acid peptide,
comprising 616-amino acids, an N-terminal extracellular domain and
a large C-terminal cytoplasmic domain, as well as a 28-amino acid
signaling peptide and a 21 amino acid short transmembrane domain
(8). RANK is primarily expressed
by cells of the macrophage/monocyte lineage, including
preosteoclastic cells, T and B cells, dendritic cells and
fibroblasts (8,22). RANK is highly expressed on the
surface of osteoclast progenitors and mature osteoclasts, which are
able to translate osteoclastogenesis signals by binding to RANKL
(6). A RANK knock-out mouse model
developed osteopetrosis and exhibited an absence of osteoclasts
(23). RANKL/RANK signaling has
been extensively studied following the discovery of the ligand and
its receptor (24,25). There are multiple pathways involved
in the transduction of RANKL/RANK signaling in osteoclastogenesis,
including nuclear factor-κB (NF-κB), c-Jun N-terminal kinase
(JNK)/activator protein 1 (AP-1), c-Myc and calcinerin/nuclear
factor of activated T-cells, cytoplasmic 1 (NFATc1) (26,27).
Three pathways participate in the activation and survival of
osteoclasts: Src, p38 and extracellular signal-regulated kinase
(ERK) (28). Adaptor molecules,
including growth factor receptor-bound protein 2 (Grb2) and TNF
receptor-associated factor (TRAF)2/5/6 also mediate RANKL/RANK
signaling (28).
3. RANKL/RANK/OPG signaling in
osteoclastogenesis
As described above, the binding of RANKL to RANK
leads to osteoclastogenesis, which may be blocked by OPG acting as
a decoy receptor for RANKL. The signaling pathways underlying
RANKL/RANK have been extensively studied, and multiple pathways are
involved in mediating RANKL/RANK signal transduction (Fig. 1).
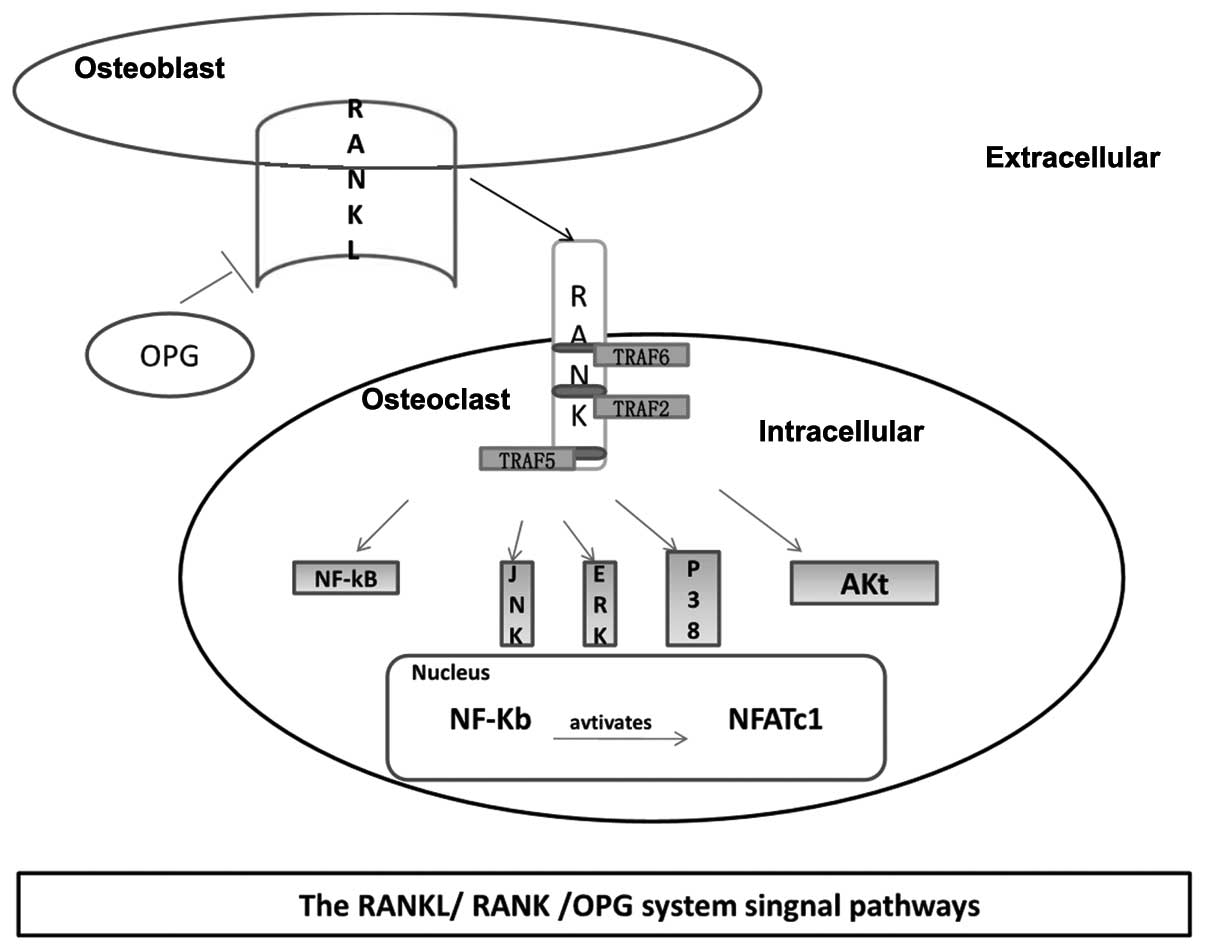 | Figure 1Major signaling pathways involved in
osteoclastogenesis. Osteoblasts produce RANKL following the binding
of RANKL to RANK on the surface of osteoclastic precursors, and
TRAF2, 5 and 6 to the RANK cytoplasmic domain. Subsequently,
multiple molecules are activated, including JNK, p38, ERK, Akt and
NF-κB. Activated NF-κB translocates into the nucleus and interacts
with NFATc1 to trigger osteoclastogenic gene transcription. TRAF,
tumor necrosis factor receptor associated factor; RANK, receptor
activator of nuclear factor-κB; RANKL, RANK ligand; JNK, c-Jun
N-terminal kinase; p38, protein 38; ERK, extracellular
signal-regulated kinase; NF-κB, nuclear factor κB; NFATc1, nuclear
factor of activated T-cells, cytoplasmic 1; OPG,
osteoprotegerin. |
RANK, like other TNF family receptors, has no
intrinsic protein kinase activating capacity to mediate signaling;
therefore, adaptor molecules are required to bind to the
intracytoplasmic domain of RANK (29). These adaptors include TNF
receptor-associated factors (TRAFs), which bind to specific sites
in the cytoplasmic domain of RANK and activate downstream molecules
(25). TRAF2, 5 and 6 are the
major TRAFS that bind to RANK; however, only TRAF6 appears to
influence RANK signaling. This is based on the observation that in
mice with deficiencies in TRAF2, 5 or 6, respectively, only
TRAF6-deficient mice developed osteopetrosis (24). Of note, two separate groups
identified osteopetrosis in TRAF6-deficient mice; one mouse model
presented with a normal number of osteoclasts which were not
active, whereas the other was found to have no osteoclasts
(30,31). The reasons underlying the
development of these two distinct phenotypes of TRAF6-deficient
mice identified have remained elusive. Another adaptor is
Grb2-associated binder (Gab) protein 2, a member of a protein
family characterized by phosphorylation at tyrosine residues, which
recruit signaling molecules containing Src homology-2 domains
(32).
A preliminary step in the induction of downstream
signaling following RANKL ligation to RANK is the binding of TRAFs
to specific RANK motifs (Motif1, -2 or -3) on the cytoplasmic
domain of RANK (33). Motif1
activates NF-κB and three mitogen-activated protein kinase pathways
(JNK, ERK and p38) in response to RANKL stimulation. The existence
of a protein complex containing TRAF6, TGF-β-activated kinase 1 and
adaptor protein TAB2 is required for the activation of these
pathways (25,33). RANKL/RANK additionally activates
the Akt/PKB pathway via a complex comprising c-Src and TRAF6 at
Motif1 (34). Therefore, following
the binding of TRAFs to RANK multiple signaling pathways are
activated. The majority of the signaling pathways associated with
osteogenesis involve NFATc1. The binding of RANKL to RANK on the
surface of osteoclast precursors recruits the adapter protein
TRAF6, resulting in the activation of NF-κB via the phosphorylation
and inactivation of inhibitory κ kinases (IKKs) and NF-κB
inhibitory kinase (25). The
binding of TRAF2 to TNFR triggers the activation of JNK;
furthermore, interleukin (IL)-1 binds to IL-1 receptor and recruits
MyD88, activating p38/ERK. These processes subsequently interact
with the NFATc1 promoter to induce NFATc1 auto-amplification of
NFATc1 and the subsequent transcription of specific genes, which
mediate the completion of the differentiation process (27,35).
The binding of TRAF6 to RANK was found to increase
intracellular calcium levels, which activated calcineurin and
dephosphorylated and activated NFATc1, resulting in its
translocation to nuclei in order to form a ternary complex with
c-Fos and c-Jun at the NFATc1 gene promoter, inducing NFATc1
expression (27). Cyclosporine A,
a calcineurin inhibitor and immunosuppressant, inhibits NFATc1
activation (36). Patients using
immunosuppressants developed bone loss, which was mainly attributed
to the inhibitory effects of immunosuppressants on osteoblasts
(36). Osteoblasts regulate
osteoclastogeneis via the expression of OPG and RANKL. The
expression of OPG in osteoblasts is regulated by certain hormones,
cytokines and the Wnt/β-catenin pathway (37). The Wnt/β-catenin pathway also
regulates bone formation and differentiation in osteoblasts. The
jagged1/notch1 pathway in osteoblasts negatively mediates
osteoclastogenesis by decreasing the number of osteoclast
progenitors and decreasing RANKL expression in stromal cells
(38). In conclusion, bone mass is
regulated by osteoclasts and osteoblasts, and at least three
pathways are involved in the regulation exerted by osteoblasts,
including RANKL/RANK, Wnt/β-catenin and jagged1/notch1.
4. RANKL/RANK/OPG system interacts with the
immune system
Bone was previously thought to be a stable inactive
organ which provided support for muscles, and protected vital
organs and hematopoietic marrow (39). However when bone modeling and
remodeling was identified, the key regulators underlying these
processes were identified to be bone resorption and formation by
osteoclasts and osteoblasts, respectively (40). Bone marrow has long been known for
its hematopoietic function in the generation of blood and immune
progenitor cells. However, further information regarding the
association between bone and the immune system remained elusive
prior to the discovery of the RANKL/RANK/OPG system. Osteoblasts,
stromal cells and T cells express RANKL and secrete M-CSF and TNF
simultaneously. The binding of RANKL and M-CSF to RANK and colony
stimulating factor 1 receptor, respectively, induces
osteoclastogenesis (41). TNF
positively influences the process by binding to its receptor. In
the immune system, T cells participate in a variety of processes
and also secrete cytokines, including TNF, IL-4 and IL-1, which are
also involved in osteoclastogenesis. Following the identification
of such associations between the immune system and bone, the field
of ‘osteoimmunology’ was created (42).
Over the past decade, rapid progress was made in the
field of osteoimmunology, elucidating the interactions and shared
mechanisms between the skeletal and immune systems. One example of
such an interaction is that between bone and the immune system in
patients with RA. RA is an autoimmune disease resulting in a
chronic, systematic inflammatory disorder, which may involve
numerous tissues and organs, but principally attacks flexible
joints causing bone loss (43). It
has been demonstrated that activated T cells, osteoblasts and
stromal cells in RA joints overexpress RANKL, leading to the
activation of osteoclasts, which enhances bone destruction
(44). Furthermore, activated T
cells may secrete cytokines, including TNF, IL1, IL17 and IL6,
which also contribute to bone loss. However, T cells also produce
anti-osteoclastogenic cytokines, including interferon (IFN)-γ and
IFN-β, which counteract the action of RANKL (26). Of note, IFN-γ and IFN-β inhibit
osteoclastogenesis via differential mechanisms. IFN-γ suppresses
osteoclastogenesis by inducing rapid degradation of TRAF6 (26). The binding of IFN-γ to IFN γ
receptor 1 recruits the signal transducer and activator of
transcription (Stat)1, resulting in the activation of Stat1.
Activated Stat1 activates the proteasome, mediating the
poly-ubiquitination of TRAF6 and therefore inducing TRAF6
degradation (26,45). By contrast, a series of in
vitro and in vivo experiments demonstrated that IFN-β
regulates osteoclastogenesis via a negative feedback mechanism
involving c-Fos, a critical factor involved in osteoclastogenesis
(46). RANKL or RANK knock-out
mice not only develop osteopetrosis, but additionally lack lymph
nodes (47), which suggests that
RANKL/RANK may influence the formation of the lymph nodes,
potentially via alterations to the function of lymph node inducer
cells. Therefore, the RANKL/RANK system is a critical regulator of
lymph node formation (48).
5. RANKL/RANK/OPG system in bone-associated
diseases
Osteoporosis
Osteoporosis has a high prevalence amongst the
elderly, in particular postmenopausal females and patients
receiving glucocorticoid treatment. The major reason underlying the
susceptibility of postmenopausal females to osteoporosis is their
characteristic lack of estrogen, which has been demonstrated to be
a significant regulator of bone density (49). Osteoporosis is characterized by
decreased bone mass and density, which may result in an increased
risk of fracture (50). Studies
have revealed a significant role of RANKL and RANK in osteoporosis,
since the number and activation of osteoclasts is responsible for
the severity of bone loss in osteoporosis (51). It has been revealed that human bone
marrow cells from untreated early postmenopausal females
demonstrated higher expression levels of RANKL in comparison to
those in an estrogen-treated group (52). Glucocorticoid-induced osteoporosis
is also mediated by the RANK/RANKL/OPG system: Glucocorticoids
stimulate RANKL expression by osteoclasts and inhibit OPG
synthesis, enhancing osteoclast differentiation and proliferation
(53).
Rheumatoid arthritis
RA is an autoimmune disease, which induces joint
inflammation and bone destruction. Within the inflamed joints,
activated T cells overexpress RANKL, thereby contributing to bone
loss (54). At the site of bone
resorption in patients with RA, synovial T cells express RANKL and
an overexpression of RANKL messenger RNA is present, which
contributes to osteoclast differentiation and activity (55). Modifying anti-rheumatic drugs which
are used in RA treatment reduce the RANKL/OPG ratio and suppress
osteoclast formation, cofirming the significance of RANKL and RANK
signaling in RA (56).
RANKL/RANK/OPG and bone heredopathia
Disorders of the RANKL/RANK/OPG system may result in
osteoporosis or osteopetrosis. Therefore, studies have been
performed in order to elucidate the mechanisms underlying bone
heredopathia, in particular heredopathia that induces osteoporosis
or osteopetrosis.
It was previously reported that two patients with
juvenile Paget’s disease, an autosomal recessive disorder
characterized by enhanced bone remodeling, osteopenia and risk of
fractures, had 100 kb homozygous deletions within the OPG gene
(57). Furthermore, idiopathic
hyperphosphatasia, an autosomal recessive bone disease
characterized by an enhanced rate of bone turnover and associated
with deformities of long bones, kyphosis and acetabular protrusions
in affected children, was found to contain a deletion in exon three
of the OPG gene (58).
Certain patients with familial Paget’s disease
presented with a mutation in exon one of RANK, which resulted in an
increase in the activation of RANK-mediated NF-κB signaling and
enhanced the number of osteoclasts and osteolysis (59). Similarly, familial expansile
osteolysis, a rare autosomal dominant disorder characterized by
focal areas of enhanced bone resorption, contains a mutation in the
signal peptide region of the RANK protein (59). In 2007, autosomal recessive
osteopetrosis, which is frequently associated with normal or
elevated numbers of non-functional osteoclasts, was demonstrated to
contain mutations in the gene coding for RANKL (60).
Bone tumors
Certain bone tumors, including multiple myeloma and
osteosarcoma, produce not only RANKL but also cytokines, which
stimulate osteolysis and bone metastasis (61,62).
Breast and prostate cancer are the most common malignant tumors
associated with bone metastasis.
The majority of patients with breast or prostate
cancer (>75%) experience skeletal complications due to bone
metastases (63). Breast cancer
cells frequently express parathyroid hormone-related protein
(PTHrP), which enhances osteoclastogenesis by increasing RANKL
expression on the surface of osteoblasts. This process may result
in osteolysis, increasing the release of growth factors, which in
turn stimulate the proliferation of tumor cells and increase the
production of PTHrP by cancer cells (64). Prostate cancer cells may express
RANK and stimulate IKKa activation, which inhibits the expression
of maspin, a known metastasis suppressor of prostate epithelial
cells (63,65). Therefore, RANKL/RANK is associated
with specific bone tumors and metastases.
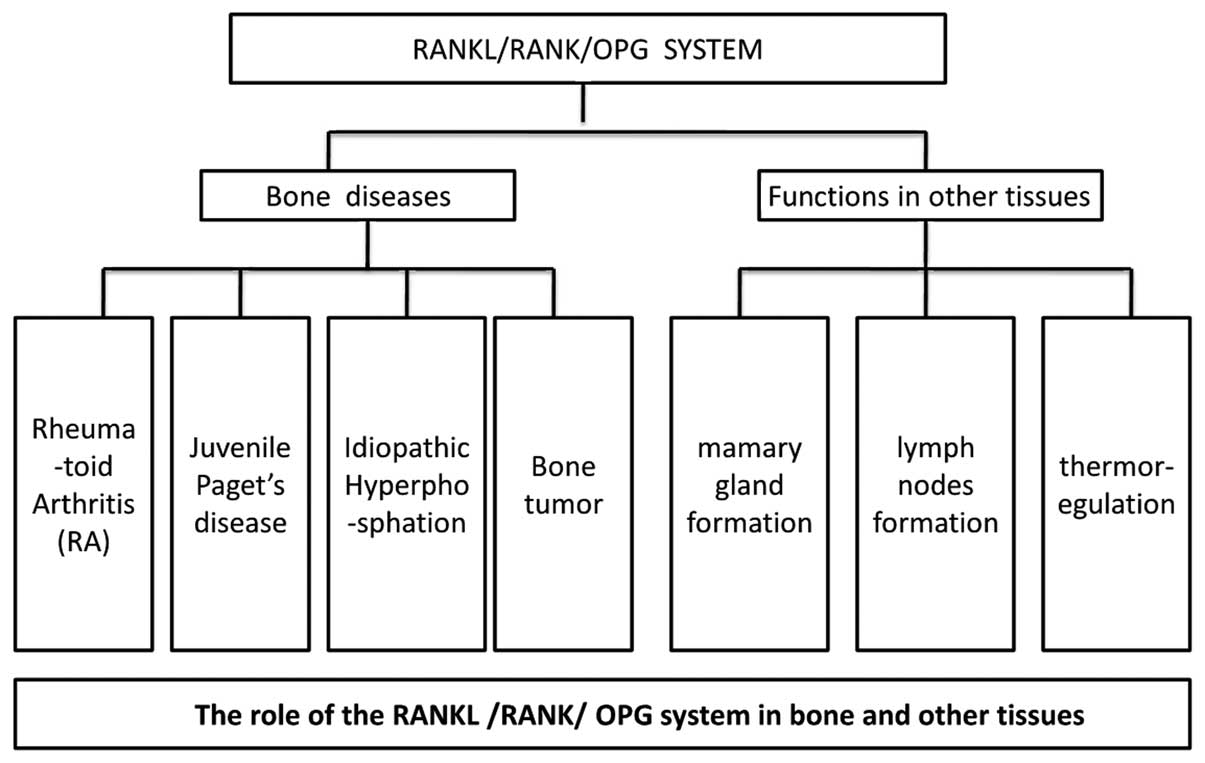
The functions of the RANKL/RANK/OPG system in
bone-associated diseases are summarized in Fig. 2.
6. RANKL/RANK/OPG system beyond bone
The discovery of the RANKL/RANK/OPG system led to
experiments involving knock-out mice deficient in RANKL, RANK or
OPG. Of note, neither RANK nor RANKL knock-out mice are able to
form alveolar mammary gland structures during pregnancy, resulting
in an absence of lactating mammary glands and leading to the death
of newborn pups (66). Further
studies indicated that RANKL expression in the mammary epithelial
cells was induced by pregnancy hormones, including prolactin,
progesterone and PTHrP. Certain RANKL/RANK downstream pathways
regulate the formation of mammary glands, including the IKKa, NF-κB
and cyclin D1 signaling pathways (67,68).
RANKL or RANK knock-out mice are not only deficient
of mammary glands but additionally lack lymph nodes (20,47).
Further studies revealed that the transfer of normal bone marrow to
RANKL or RANK knock-out mice did not rescue lymph-node formation,
which suggested that the RANKL mutant lymphocytes were not the main
cause underlying the defective lymph nodes (48). However, the precise mechanism
underlying this phenomenon has remained elusive.
RANKL and RANK are also expressed in tissues other
than bone, including lung, kidney, spleen, thymus and mammary
glands (69,70). The function of RANKL/RANK in the
brain was not elucidated until 2009 (9). RANKL/RANK is localized to the lateral
septal nucleus of the hypothalamus in the brain, an area associated
with thermoregulation, suggesting a potential role for RANKL/RANK
in mediating thermoregulation (9).
A series of experiments were performed, which revealed that
injection of recombinant RANKL into mice resulted in marked
hyperthermia and these RANKL-induced phenomena were attenuated
following pre-treatment with OPG (9). By contrast, peripheral
intraperitoneal injection of RANKL did not alter body temperature
nor activity (9). Therefore,
central but not peripheral RANKL/RANK signaling is required to
induce hyperthermia. The functions of the RANKL/RANK/OPG system
beyond bones are summarized in Fig.
2.
7. Conclusion
The discovery of RANKL/RANK/OPG in the 1990s was a
breakthrough in the elucidation of the biology of bone resorption
and formation. Disorders of the RANKL/RANK/OPG system result in a
variety of diseases, including osteoporosis and RA. RANKL/RANK
signaling not only has a significant role in bone but also
functions in other tissues. The system regulates lymph-node
formation, mammary-gland development, fever control and certain
metastatic tumors.
Since the RANKL/RANK/OPG system functions in
numerous tissues and its disorder is associated with multiple
diseases, it presents a significant potential therapeutic target. A
series of pharmacological experiments have been performed, the
results of which indicated that OPG- and RANK-Fc inhibited bone
loss in models of sex-steroid deficiency, glucocorticoid-induced
osteoporosis, RA, multiple myeloma and metastatic bone disease
(71,72). However, OPG- and RANK-Fc have not
been taken to further clinical trial stages due to concerns
regarding potential side effects on the immune system (43). A promising drug targeting the
RANKL/RANK/OPG system is denosumab (73), which is a monoclonal antibody
against RANKL. Phase II and III clinical trials of denosumab have
not identified any significant side effects (74,75).
Furthermore, curative effects have been observed in patients with
various bone disorders, including postmenopausal osteoporosis, RA,
multiple myeloma and metastatic bone disease (72). However, whether the long-term
inhibition of RANKL has any adverse effects remains to be
elucidated.
References
|
1
|
Raggatt LJ and Partridge NC: Cellular and
molecular mechanisms of bone remodeling. J Biol Chem.
285:25103–25108. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Boyce BF, Xing L, Shakespeare W, Wang Y,
Dalgarno D, Iuliucci J and Sawyer T: Regulation of bone remodeling
and emerging breakthrough drugs for osteoporosis and osteolytic
bone metastases. Kidney Int. 63:S2–S5. 2003. View Article : Google Scholar
|
|
3
|
van Oers RF, Ruimerman R, Tanck E, et al:
A unified theory for osteonal and hemi-osteonal remodeling. Bone.
42:250–259. 2008. View Article : Google Scholar
|
|
4
|
Boyce BF and Xing L: Functions of
RANKL/RANK/OPG in bone modeling and remodeling. Arch Biochem
Biophys. 473:139–146. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Greenfield EM, Bi Y and Miyauchi A:
Regulation of osteoclast activity. Life Sci. 65:1087–1102. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lacey D, Timms E, Tan H-L, Kelley M,
Dunstan C, Burgess T, Elliott R, Colombero A, Elliott G and Scully
S: Osteoprotegerin ligand is a cytokine that regulates osteoclast
differentiation and activation. Cell. 93:165–176. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Simonet W, Lacey D, Dunstan C, Kelley M,
Chang M-S, Lüthy R, Nguyen H, Wooden S, Bennett L and Boone T:
Osteoprotegerin: a novel secreted protein involved in the
regulation of bone density. Cell. 89:309–319. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Anderson DM, Maraskovsky E, Billingsley
WL, Dougall WC, Tometsko ME, Roux ER, Teepe MC, DuBose RF, Cosman D
and Galibert L: A homologue of the TNF receptor and its ligand
enhance T-cell growth and dendritic-cell function. Nature.
390:175–179. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hanada R, Leibbrandt A, Hanada T, Kitaoka
S, Furuyashiki T, Fujihara H, Trichereau J, Paolino M, Qadri F, et
al: Central control of fever and female body temperature by
RANKL/RANK. Nature. 462:505–509. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Karsenty G and Wagner EF: Reaching a
genetic and molecular understanding of skeletal development. Dev
Cell. 2:389–406. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yasuda H, Shima N, Nakagawa N, Yamaguchi
K, Kinosaki M, Mochizuki S-i, Tomoyasu A, Yano K, Goto M, et al:
Osteoclast differentiation factor is a ligand for
osteoprotegerin/osteoclastogenesis-inhibitory factor and is
identical to TRANCE/RANKL. Proc Natl Acad Sci USA. 95:3597–3602.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wong BR, Josien R, Lee SY, et al: TRANCE
(tumor necrosis factor [TNF]-related activation-induced cytokine),
a new TNF family member predominantly expressed in T cells, is a
dendritic cell-specific survival factor. J Exp Med. 186:2075–2080.
1997. View Article : Google Scholar
|
|
13
|
Rodan GA and Martin TJ: Role of
osteoblasts in hormonal control of bone resorption - a hypothesis.
Calcif Tissue Int. 33:349–351. 1981. View Article : Google Scholar
|
|
14
|
Yasuda H, Shima N, Nakagawa N, Mochizuki
S-I, Yano K, Fujise N, Sato Y, Goto M, Yamaguchi K and Kuriyama M:
Identity of osteoclastogenesis inhibitory factor (OCIF) and
osteoprotegerin (OPG): a mechanism by which OPG/OCIF inhibits
osteoclastogenesis in vitro. Endocrinology. 139:1329–1337.
1998.PubMed/NCBI
|
|
15
|
Tat SK, Pelletier J-P, Velasco CR,
Padrines M and Martel-Pelletier J: New perspective in
osteoarthritis: the OPG and RANKL system as a potential therapeutic
target? Keio J Med. 58:29–40. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Caetano-Lopes J, Canhao H and Fonseca JE:
Osteoblasts and bone formation. Acta Reumatol Port.
32:1032007.PubMed/NCBI
|
|
17
|
Boyce BF and Xing L: Biology of RANK,
RANKL, and osteoprotegerin. Arthritis Res Ther. 9:S12007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lum L, Wong BR, Josien R, Becherer JD,
Erdjument-Bromage H, Schlöndorff J, Tempst P, Choi Y and Blobel CP:
Evidence for a role of a tumor necrosis factor-α (TNF-α)-converting
enzyme-like protease in shedding of TRANCE, a TNF family member
involved in osteoclastogenesis and dendritic cell survival. J Biol
Chem. 274:13613–13618. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wong BR, Rho J, Arron J, Robinson E,
Orlinick J, Chao M, Kalachikov S, Cayani E, Bartlett FS and Frankel
WN: TRANCE is a novel ligand of the tumor necrosis factor receptor
family that activates c-Jun N-terminal kinase in T cells. J Biol
Chem. 272:25190–25194. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kong Y-Y, Yoshida H, Sarosi I, Tan H-L,
Timms E, Capparelli C, Morony S, Oliveira-dos-Santos AJ, Van G and
Itie A: OPGL is a key regulator of osteoclastogenesis, lymphocyte
development and lymph-node organogenesis. Nature. 397:315–323.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Haynes DR, Barg E, Crotti TN, Holding C,
Weedon H, Atkins GJ, Zannetino A, Ahern M, Coleman M and
Roberts-Thomson PJ: Osteoprotegerin expression in synovial tissue
from patients with rheumatoid arthritis, spondyloarthropathies and
osteoarthritis and normal controls. Rheumatology (Oxford).
42:123–134. 2003. View Article : Google Scholar
|
|
22
|
Hsu H, Lacey DL, Dunstan CR, Solovyev I,
Colombero A, Timms E, Tan HL, Elliott G, Kelley MJ and Sarosi I:
Tumor necrosis factor receptor family member RANK mediates
osteoclast differentiation and activation induced by
osteoprotegerin ligand. Proc Natl Acad Sci USA. 96:3540–3545. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li J, Sarosi I, Yan X-Q, Morony S,
Capparelli C, Tan H-L, McCabe S, Elliott R, Scully S and Van G:
RANK is the intrinsic hematopoietic cell surface receptor that
controls osteoclastogenesis and regulation of bone mass and calcium
metabolism. Proc Natl Acad Sci USA. 97:1566–1571. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Galibert L, Tometsko ME, Anderson DM, et
al: The involvement of multiple tumor necrosis factor receptor
(TNFR)-associated factors in the signaling mechanisms of receptor
activator of NF-kappaB, a member of the TNFR superfamily. J Biol
Chem. 273:34120–34127. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ye H, Arron JR, Lamothe B, Cirilli M,
Kobayashi T, Shevde NK, Segal D, Dzivenu OK, Vologodskaia M, et al:
Distinct molecular mechanism for initiating TRAF6 signalling.
Nature. 418:443–447. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Takayanagi H, Ogasawara K, Hida S, Chiba
T, Murata S, Sato K, Takaoka A, Yokochi T, Oda H, et al:
T-cell-mediated regulation of osteoclastogenesis by signalling
cross-talk between RANKL and IFN-gamma. Nature. 408:600–605. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Takayanagi H, Kim S, Koga T, Nishina H,
Isshiki M, Yoshida H, Saiura A, Isobe M, Yokochi T, et al:
Induction and activation of the transcription factor NFATc1 (NFAT2)
integrate RANKL signaling in terminal differentiation of
osteoclasts. Dev Cell. 3:889–901. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kim H-H, Lee DE, Shin JN, Lee YS, Jeon YM,
Chung C-H, Ni J, Kwon BS and Lee ZH: Receptor activator of NF-κB
recruits multiple TRAF family adaptors and activates c-Jun
N-terminal kinase. FEBS Lett. 443:297–302. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Darnay BG, Haridas V, Ni J, et al:
Characterization of the intracellular domain of receptor activator
of NF-kappaB (RANK). Interaction with tumor necrosis factor
receptor-associated factors and activation of NF-kappab and c-Jun
N-terminal kinase. J Biol Chem. 273:20551–20555. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Naito A, Azuma S, Tanaka S, Miyazaki T,
Takaki S, Takatsu K, Nakao K, Nakamura K, Katsuki M and Yamamoto T:
Severe osteopetrosis, defective interleukin-1 signalling and lymph
node organogenesis in TRAF6-deficient mice. Genes Cells. 4:353–362.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lomaga MA, Yeh W-C, Sarosi I, Duncan GS,
Furlonger C, Ho A, Morony S, Capparelli C, Van G and Kaufman S:
TRAF6 deficiency results in osteopetrosis and defective
interleukin-1, CD40, and LPS signaling. Genes Dev. 13:1015–1024.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wada T, Nakashima T, Oliveira-dos-Santos
AJ, Gasser J, Hara H, Schett G and Penninger JM: The molecular
scaffold Gab2 is a crucial component of RANK signaling and
osteoclastogenesis. Nat Med. 11:394–399. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu W, Xu D, Yang H, Xu H, Shi Z, Cao X,
Takeshita S, Liu J, Teale M and Feng X: Functional identification
of three receptor activator of NF-κB cytoplasmic motifs mediating
osteoclast differentiation and function. J Biol Chem.
279:54759–54769. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wong BR, Besser D, Kim N, Arron JR,
Vologodskaia M, Hanafusa H and Choi Y: TRANCE, a TNF family member,
activates Akt/PKB through a signaling complex involving TRAF6 and
c-Src. Mol Cell. 4:1041–1049. 1999. View Article : Google Scholar
|
|
35
|
Asagiri M, Sato K, Usami T, Ochi S,
Nishina H, Yoshida H, Morita I, Wagner EF, Mak TW and Serfling E:
Autoamplification of NFATc1 expression determines its essential
role in bone homeostasis. J Exp Med. 202:1261–1269. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Koga T, Matsui Y, Asagiri M, Kodama T, de
Crombrugghe B, Nakashima K and Takayanagi H: NFAT and Osterix
cooperatively regulate bone formation. Nat Med. 11:880–885. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Theoleyre S, Wittrant Y, Tat SK, Fortun Y,
Redini F and Heymann D: The molecular triad OPG/RANK/RANKL:
involvement in the orchestration of pathophysiological bone
remodeling. Cytokine Growth Factor Rev. 15:457–475. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bai S, Kopan R, Zou W, Hilton MJ, Ong C-t,
Long F, Ross FP and Teitelbaum SL: NOTCH1 regulates
osteoclastogenesis directly in osteoclast precursors and indirectly
via osteoblast lineage cells. J Biol Chem. 283:6509–6518. 2008.
View Article : Google Scholar
|
|
39
|
Seeman E and Delmas PD: Bone quality-the
material and structural basis of bone strength and fragility. N
Engl J Med. 354:2250–2261. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Takayanagi H: Osteoimmunology: shared
mechanisms and crosstalk between the immune and bone systems. Nat
Rev Immunol. 7:292–304. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Roodman GD: Cell biology of the
osteoclast. Exp Hematol. 27:1229–1241. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Walsh MC, Kim N, Kadono Y, et al:
Osteoimmunology: interplay between the immune system and bone
metabolism. Annu Rev Immunol. 24:33–63. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Takayanagi H: New developments in
osteoimmunology. Nat Rev Rheumatol. 8:684–689. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Takayanagi H: Osteoimmunology and the
effects of the immune system on bone. Nat Rev Rheumatol. 5:667–676.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Schroder K, Hertzog PJ, Ravasi T and Hume
DA: Interferon-γ: an overview of signals, mechanisms and functions.
J Leukoc Biol. 75:163–189. 2004. View Article : Google Scholar
|
|
46
|
Takayanagi H, Kim S, Matsuo K, Suzuki H,
Suzuki T, Sato K, Yokochi T, Oda H, Nakamura K and Ida N: RANKL
maintains bone homeostasis through c-Fos-dependent induction of
interferon-β. Nature. 416:744–749. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Dougall WC, Glaccum M, Charrier K,
Rohrbach K, Brasel K, De Smedt T, Daro E, Smith J, Tometsko ME and
Maliszewski CR: RANK is essential for osteoclast and lymph node
development. Genes Dev. 13:2412–2424. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kim D, Mebius RE, MacMicking JD, Jung S,
Cupedo T, Castellanos Y, Rho J, Wong BR, Josien R and Kim N:
Regulation of peripheral lymph node genesis by the tumor necrosis
factor family member TRANCE. J Exp Med. 192:1467–1478. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Raisz LG: Pathogenesis of osteoporosis:
concepts, conflicts, and prospects. J Clin Invest. 115:3318–3325.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ghannam NN: Book review: Assessment of
fracture risk and its application to screening for postmenopausal
osteoporosis. Ann Saudi Med. 14:5271994.PubMed/NCBI
|
|
51
|
Ominsky MS, Li X, Asuncion FJ, Barrero M,
Warmington KS, Dwyer D, Stolina M, Geng Z, Grisanti M and Tan HL:
RANKL inhibition with osteoprotegerin increases bone strength by
improving cortical and trabecular bone architecture in
ovariectomized rats. J Bone Mine Res. 23:672–682. 2008. View Article : Google Scholar
|
|
52
|
Cummings SR, Martin JS, McClung MR, Siris
ES, Eastell R, Reid IR, Delmas P, Zoog HB, Austin M and Wang A:
Denosumab for prevention of fractures in postmenopausal women with
osteoporosis. N Engl J Med. 361:756–765. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Vega D, Maalouf NM and Sakhaee K: The role
of receptor activator of nuclear factor-κB (RANK)/RANK
ligand/osteoprotegerin: clinical implications. J Clin Endocrinol
Metab. 92:4514–4521. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Pettit A, Walsh N, Manning C, Goldring S
and Gravallese E: RANKL protein is expressed at the pannus-bone
interface at sites of articular bone erosion in rheumatoid
arthritis. Rheumatology (Oxford). 45:1068–1076. 2006. View Article : Google Scholar
|
|
55
|
Ainola M, Mandelin J, Liljestrom M,
Konttinen Y and Salo J: Imbalanced expression of RANKL and
osteoprotegerin mRNA in pannus tissue of rheumatoid arthritis. Clin
Exp Rheumatol. 26:2402008.PubMed/NCBI
|
|
56
|
Haynes D, Crotti T, Weedon H, Slavotinek
J, Au V, Coleman M, Roberts-Thomson PJ, Ahern M and Smith MD:
Modulation of RANKL and osteoprotegerin expression in synovial
tissue from patients with rheumatoid arthritis in response to
disease-modifying antirheumatic drug treatment and correlation with
radiologic outcome. Arthritis Rheum. 59:911–920. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Whyte MP, Obrecht SE, Finnegan PM, Jones
JL, Podgornik MN, McAlister WH and Mumm S: Osteoprotegerin
deficiency and juvenile Paget’s disease. N Engl J Med. 347:175–184.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Cundy T, Hegde M, Naot D, Chong B, King A,
Wallace R, Mulley J, Love DR, Seidel J and Fawkner M: A mutation in
the gene TNFRSF11B encoding osteoprotegerin causes an idiopathic
hyperphosphatasia phenotype. Hum Mol Genet. 11:2119–2127. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Hughes AE, Ralston SH, Marken J, Bell C,
MacPherson H, Wallace RG, van Hul W, Whyte MP, Nakatsuka K and Hovy
L: Mutations in TNFRSF11A, affecting the signal peptide of RANK,
cause familial expansile osteolysis. Nat Genet. 24:45–48. 2000.
View Article : Google Scholar
|
|
60
|
Sobacchi C, Frattini A, Guerrini MM,
Abinun M, Pangrazio A, Susani L, Bredius R, Mancini G, Cant A and
Bishop N: Osteoclast-poor human osteopetrosis due to mutations in
the gene encoding RANKL. Nat Genet. 39:960–962. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Mori K, Le Goff B, Berreur M, Riet A,
Moreau A, Blanchard F, Chevalier C, Guisle-Marsollier I, Leger J
and Guicheux J: Human osteosarcoma cells express functional
receptor activator of nuclear factor-kappa B. J Pathol.
211:555–562. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Giuliani N, Colla S, Sala R, Moroni M,
Lazzaretti M, La Monica S, Bonomini S, Hojden M, Sammarelli G and
Barillè S: Human myeloma cells stimulate the receptor activator of
nuclear factor-κB ligand (RANKL) in T lymphocytes: a potential role
in multiple myeloma bone disease. Blood. 100:4615–4621. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Mundy GR: Metastasis: Metastasis to bone:
causes, consequences and therapeutic opportunities. Nat Rev Cancer.
2:584–593. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
64
|
Weigelt B, Peterse JL and van’t Veer LJ:
Breast cancer metastasis: markers and models. Nat Rev Cancer.
5:591–602. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Dougall WC and Chaisson M: The
RANK/RANKL/OPG triad in cancer-induced bone diseases. Cancer
Metastasis Rev. 25:541–549. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Fata JE, Kong Y-Y, Li J, Sasaki T,
Irie-Sasaki J, Moorehead RA, Elliott R, Scully S, Voura EB and
Lacey DL: The osteoclast differentiation factor
osteoprotegerin-ligand is essential for mammary gland development.
Cell. 103:41–50. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Srivastava S, Matsuda M, Hou Z, Bailey JP,
Kitazawa R, Herbst MP and Horseman ND: Receptor activator of NF-κB
ligand induction via Jak2 and Stat5a in mammary epithelial cells. J
Biol Chem. 278:46171–46178. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Cao Y, Bonizzi G, Seagroves TN, Greten FR,
Johnson R, Schmidt EV and Karin M: IKKα provides an essential link
between RANK signaling and cyclin D1 expression during mammary
gland development. Cell. 107:763–775. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Nakagawa N, Kinosaki M, Yamaguchi K, Shima
N, Yasuda H, Yano K, Morinaga T and Higashio K: RANK is the
essential signaling receptor for osteoclast differentiation factor
in osteoclastogenesis. Biochem Biophys Res Commun. 253:395–400.
1998. View Article : Google Scholar
|
|
70
|
Kartsogiannis V, Zhou H, Horwood N, Thomas
R, Hards D, Quinn J, Niforas P, Ng K, Martin T and Gillespie M:
Localization of RANKL (receptor activator of NFκB ligand) mRNA and
protein in skeletal and extraskeletal tissues. Bone. 25:525–534.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Chau D, Becker DL, Coombes ME, et al:
Cost-effectiveness of denosumab in the treatment of postmenopausal
osteoporosis in Canada. J Med Econ. 15(Suppl 1): 3–14. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Dempster DW, Lambing CL, Kostenuik PJ and
Grauer A: Role of RANK ligand and denosumab, a targeted RANK ligand
inhibitor, in bone health and osteoporosis: a review of preclinical
and clinical data. Clin Ther. 34:521–536. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Lacey DL, Boyle WJ, Simonet WS, et al:
Bench to bedside: elucidation of the OPG–RANK–RANKL pathway and the
development of denosumab. Nat Rev Drug Discov. 11:401–419. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Lipton A, Fizazi K, Stopeck AT, et al:
Superiority of denosumab to zoledronic acid for prevention of
skeletal-related events: a combined analysis of 3 pivotal,
randomised, phase 3 trials. Eur J Cancer. 48:3082–3092. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
McClung MR, Lewiecki EM, Geller ML, et al:
Effect of denosumab on bone mineral density and biochemical markers
of bone turnover: 8-year results of a phase 2 clinical trial.
Osteoporos Int. 24:227–235. 2013. View Article : Google Scholar :
|