Introduction
Taxol (paclitaxel) has emerged as an essential
chemotherapeutic agent for the treatment of multiple types of
tumor, including ovarian, prostate and non-small cell lung cancer
(1–3). Taxol stabilizes the structure of
microtubules by disrupting the dynamic equilibrium between soluble
tubulin dimers and their polymerized form (3), therefore, cells treated with
paclitaxel have problems with spindle assembly, cell division and
chromosome segregation (3). Taxol
is a potent anticancer drug, causing cell cycle arrest in the late
G2 or mitotic phases (4). However,
a significant percentage of patients develop drug resistance during
the course of treatment with Taxol, and the emergence of
drug-resistant cancer cells has limited its clinical efficacy
(3,5). Therefore, it is imperative to develop
novel strategies to reduce or overcome chemoresistance in cancer.
The mechanism underlying Taxol resistance has been widely
investigated. The overexpression of anti-apoptotic proteins,
including survivin (6), myeloid
cell leukemia 1 (7) and B cell
lymphoma-2 (8), has been
identified as an underlying mechanism contributing to the
acquisition of Taxol resistance. In addition, evidence suggests
that multiple upregulated oncogenes, including epidermal growth
factor receptor 2 (ERBB2) (9,10),
Akt (11) and sarcoma (12), may be directly associated with drug
resistance in cancer.
Cancer cells exhibit an increased dependency on
aerobic glycolysis, fatty acid synthesis and glutaminolysis for
proliferation (13).
Glutaminolysis is the conversion of glutamine to glutamate, which
is the first, and a rate-limiting step of glutamine utilization
catalyzed by glutaminase (GLS). There are two isoforms of GLS:
GLS1, which has been reported to be expressed in non-hepatic types
of human tissue and tumor (14),
and GLS2, a liver isoform, which is involved in the urea cycle
(15). Glutaminolysis is not only
an important procedure for providing adenosine triphosphate and the
reducing equivalent, but is also utilized to meet biosynthetic,
energetic and reductive needs for highly proliferating cells
(16). It has been reported that
cancer cells are particularly sensitive to glutamine deprivation
and are unable to proliferate in culture in its absence (17).
The present study investigated the glutamine
metabolism of breast cancer cells following treatment with Taxol
in vitro. The glutamine uptake and the expression of GLS1 in
breast cancer cells was measured in response to various
concentrations of Taxol treatment. A Taxol-resistant cancer cell
line was established and used to investigate the roles of GLS1 in
Taxol-resistance through the regulation of glutamine metabolism. In
addition, the present study aimed to determine whether GLS1 may be
the therapeutic target for overcoming Taxol resistance, and to
provide a novel insight into the cellular and molecular mechanisms
involved in Taxol-resistant breast cancer.
Materials and methods
Cells and cell culture
The MDA-MB-231 and BT474 human breast cancer cell
lines were purchased from American Type Culture Collection
(Manassas, VA, USA). All cells were cultured in Dulbecco’s modified
Eagle’s medium (DMEM)/F-12 (Mediatech, Inc., Manassas, VA, USA),
supplemented with 10% fetal bovine serum (FBS; Life Technologies,
Carlsbad, CA, USA) and 10% penicillin/streptomycin (Life
Technologies). The cells were cultured at 37°C in a humidified
incubator with 95% air and 5% CO2.
Antibodies and reagents
Mouse anti-GLS monoclonal antibody was purchased
from Abcam (1:100; ab60709; Cambridge, MA, USA) and rabbit
anti-β-actin polyclonal antibody was purchased from Cell Signaling
Technology, Inc. (1:2,000; cat. no. #4967; Danvers, MA, USA). A
vector containing the wild-type open reading frame clone of the
Homo sapiens protein, GLS1, was purchased from OriGene
Technologies (cat. no. RC206265; Rockville, MD, USA). The siGLS1
and control siRNA were purchased from Ambion Life Technologies
(Austin, TX, USA). Taxol was purchased from Sigma-Aldrich (St.
Louis, MO, USA).
Generation of a Taxol-resistant cell
line
The MDA-MB-231 cells (1×106/10 cm dish)
were treated with gradually increasing concentrations of Taxol (50,
100, 150 and 200 nM) in regular cell culture conditions (DMEM/F-12
supplemented with 10% FBS and 10% penicillin/streptomycin) for the
selection of resistant cells. The medium containing taxol was
refreshed every 4 days for 2 months, and then several resistant
cell clones were developed from the parental cell line. The
Taxol-resistant cell clones were pooled and used for subsequent
experiments. Following selection and during the culture/passage of
cells, the cells were treated with 200 nM Taxol each month in order
to maintain Taxol resistance.
Plasmid DNA and siRNA transfections
The transfections were performed using
Oligofectamine™ Transfection reagent (Invitrogen Life Technologies,
Carlsbad, CA, USA), according to the manufacturer’s instructions.
Briefly, between 0.5 and 1×106 cells were plated into
6-well plates overnight at 37°C in a humidified incubator with 95%
air and 5% CO2 to reach between 70 and 90% confluence.
The following day, the plasmid DNA (4 μg) or siRNA (100 nM)
were diluted in Opti-MEM® I Reduced Serum medium (Life
Technologies). The diluted DNA or siRNA was then mixed with
Oligofectamine™ for the formation of DNA/siRNA-lipid complexes in a
total volume of 250 μl Opti-MEM® I Reduced Serum medium.
After 10–15 min incubation at room temperature, the mixture was
added in to the cell medium (DMEM/F-12 supplemented with 10% FBS
and 10% penicillin/streptomycin) and the cells were incubated for
48 h at 37°C in a humidified incubator with 95% air and 5%
CO2. Subsequently, whole-cell lysates were prepared for
further analysis using Novex® NP40 Cell Lysis Buffer
(Invitrogen Life Technologies), as previously described (7).
Cell viability assays
A total of 1×104 cells were seeded into
each well of a 48-well plate and incubated overnight. The medium
was replaced with fresh medium, with or without Taxol at various
concentrations (MDA-MB-231 cells: 0, 5, 10, 20, 40, 80, 120, 160
and 200 nM; BT-474 cells: 0, 0.05, 0.1, 0.2, 0.4, 0.8, 1.2, 1.6,
2.0 and 2.4 μM) and incubated for 48 h at 37°C in a
humidified incubator with 95% air and 5% CO2. The cell
viability was measured using a 3-(4,5
dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay (Life
Technologies). The absorbance was measured spectrophotometrically
at 570 nm using a Universal Microplate Reader EL800 (Bio-Tek
instruments, Inc., Vermont, MA, USA).
Glutamine uptake assay
A glutamine uptake assay was performed using a
Glutamine and Glutamate Determination kit (GLN1-1KT;
Sigma-Aldrich), according to the manufacturer’s instructions.
Briefly, MDA-MB-231 and BT474 cells with or without taxol
treatments; MDA-MB-231 parental and taxol resistant cells; BT474
siGLS1 and control siRNA cells (2×105/well) were plated
into 6-well plates for 24 h at 37°C in a humidified incubator with
95% air and 5% CO2. The cell lysates were collected and
10 μg total protein of each sample was diluted to 250
μl, the cell-free supernatant samples were collected and
analyzed in triplicate using the glutamine uptake assay kits. The
absorbance was measured at 340 nm for glutamine in a 96-well
plate-reader (SpectraMax M2 spectrophotometer; Molecular Devices,
Sunnyvale, CA, USA). All values were normalized to the total
protein from each well.
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR)
The total RNA from MDA-MB-231 and BT474 cells under
taxol treatments and the taxol-resistant and parental MDA-MB-231
cells was extracted following homogenization of the cells and
tissues (QIAshredder; cat. no. 79656; Qiagen, Germantown, MD, USA)
using an RNeasy Mini kit (Qiagen) and performing DNase digestion
(RNase free DNase kit; Qiagen) during the RNA extraction. The total
RNA (1 μg) was reverse transcribed using a High Capacity
cDNA Reverse Transcription kit (Applied Biosystems, Foster City,
CA, USA). The cDNA reaction was diluted to 1:10 with
ddH2O for use as the template for qPCR. TaqMan Gene
Expression Assays primers (Life Technologies) and GLS1 specific
probes were used for expression analysis and primers and probes
against 18S ribosomal RNA (Applied Biosystems) were used as
internal controls. The qPCR amplifications were performed in a
final reaction volume of 10 μl containing, 5.5 μl
TaqMan Universal PCR Master mix (Applied Biosystems), 0.5 μl
primers and probes mix and 4.5 μg cDNA diluted solution.
Primer sequences used were as follows: 18S rRNA forward,
5′-TGCTGTCCCTGTATGCCTCT-3′ and reverse, 5′-TGTAGCCACGCTCGGTCA-3′.
The cycling conditions were as follows: 2 min at 50°C, 10 min at
95°C, 40 cycles of denaturation for 15 sec at 95°C and
annealing/extension for 1 min at 60°C. The reactions were performed
using the Step 1 Plus Real-Time PCR system thermocycler (Applied
Biosystems) and all qPCR reactions were performed in triplicate and
repeated at least twice. The comparative threshold cycle (Ct) for
the mRNA expression was calculated relative to the Ct of 18S
ribosomal RNA. The relative mRNA expression was calculated using
the 2−ΔΔCt method (7).
The GLS1 primers used for qPCR were as follows: Forward
5′-CTTTCCATGTTGGTCTTCC-3′ and reverse
5′-AAACAAGATCGTGACAAAAGTGAA-3′.
Western blot analysis
The cells (5×105/300 μl lysis
buffer) were lysed in 1X SDS sample buffer. The proteins (40
μg/well) were subsequently resolved by electrophoresis using
SDS-PAGE (Novex® 4–20% Tris-Glycine Mini Gels; Life
Technologies) and transferred onto nitrocellulose membranes (Life
Technologies). The membranes were probed with primary antibodies
overnight at 37°C, followed by incubating with the appropriate
horseradish peroxidase-conjugated secondary antibodies for 2 h at
room temperature prior to detection using a Super Signal Enhanced
Chemiluminescence kit (Pierce Biotechnology, Inc., Rockford, IL,
USA). For sequential blotting, the membranes were stripped using
Restore Western Blot Stripping Buffer (Pierce Biotechnology, Inc.)
and re-probed with the appropriate antibodies.
Statistical analysis
Statistical analysis was performed using unpaired
Student’s t-test with GraphPad Prism 5.0 software (GraphPad
Software, Inc., La Jolla, CA, USA). The data are expressed as the
mean and P<0.05 was considered to indicate a statistically
significant difference.
Results
Glutamine metabolism and the expression
of GLS1 are induced by treatment with Taxol
A previous study demonstrated that Taxol-resistant
breast cancer cells exhibited elevated levels of glucose metabolism
(18), suggesting that there is an
association between cellular metabolism and Taxol-induced cell
apoptosis. The present study examined whether treatment with Taxol
regulated glutamine metabolism in breast cancer cells. Notably, a
significant upregulation of glutamine metabolism was observed
following treatment with Taxol at multiple concentrations (Fig. 1A). The MDA-MB-231 and BT-474
(Fig. 1A) cells exhibited an
increased glutamine uptake following treatment with Taxol at
low-toxic concentrations (MDA-MB-231: 5, 10 and 15 nM; BT474: 10,
20 and 40 nM) for 48 h. Since the first stage of glutamine
metabolism requires GLS1 to generate glutamate (15), the expression of GLS1 following
treatment with Taxol was assessed. The results revealed that the
protein and mRNA expression of GLS1 were significantly upregulated
by low-toxic treatment with Taxol (Fig. 1B and C). This suggested that
dysregulated glutamine metabolism and GLS1 may be therapeutic
targets for Taxol-induced cancer cell apoptosis.
Taxol-resistant breast cancer cells
exhibit upregulated glutamine metabolism and expression of
GLS1
To investigate the mechanisms underlying
Taxol-induced glutamine metabolism in breast cancer cells,
Taxol-resistant cells originating from MDA-MB-231 were isolated by
gradually treating parental cells with increasing concentrations of
Taxol for 8 weeks. The Taxol-resistant MDA-MB-231 colonies were
pooled as the Taxol-resistant cells. As shown in Fig. 2A, the Taxol-resistant MDA-MB-231
cells were insensitive to regular Taxol treatments compared with
the parental cells. The half maximal inhibitory concentration
(IC50) of the parental cells was ~100 nM, while the
IC50 of the Taxol-resistant cells was increased to ~300
nM. The glutamine metabolism in Taxol-sensitive and resistant cells
were compared. The glutamine uptake was significantly upregulated
in the Taxol-resistant cells compared with the Taxol-sensitive
cells (Fig. 2B). Consistently, the
protein and mRNA expression levels of GLS1 were upregulated in the
Taxol-resistant cells (Fig. 2C and
D). These findings suggested that upregulated glutamine metabolism
may be a possible mechanism underlying Taxol resistance in breast
cancer cells.
Exogenous overexpression of GLS1 renders
breast cancer cells resistant to Taxol
The results suggested that GLS1 may be involved in
the Taxol resistance exhibited by breast cancer cells. To further
examine the function of GLS1 in Taxol resistance, an overexpression
vector, containing WT GLS1, was transiently transfected into the
MDA-MB-231 and BT-474 cells (Fig.
3A and B). The sensitivities of the cells overexpressing GLS1
following treatment with Taxol were compared with the empty
vector-transfected cells. Notably, the breast cancer cells
overexpressing GLS1 exhibited increased resistance to treatment
with Taxol compared with the control cells (Fig. 3A and B). The MDA-MB-231 cells
overexpressing GLS1 demonstrated an IC50 of ~200 nM,
compared with control cells, with an IC50 of 100 nM.
Similarly, the IC50 of the BT-474 control cells was 1.2
μM, while the IC50 of the Taxol-resistant BT-474
cells was 2.2 μM. These results further supported that
upregulated expression of GSL1 contributed to Taxol resistance in
breast cancer cells.
Inhibition of GLS1 resensitizes
Taxol-resistant cancer cells to Taxol
The results revealed that treatment with Taxol
induced glutamine metabolism and that Taxol-resistant breast cancer
cells exhibited upregulated glutamine metabolism and expression of
GLS1. The present study, therefore, hypothesized that the
combination of GLS1 inhibition and treatment with Taxol may exhibit
a synergistic effect to overcome Taxol resistance through the
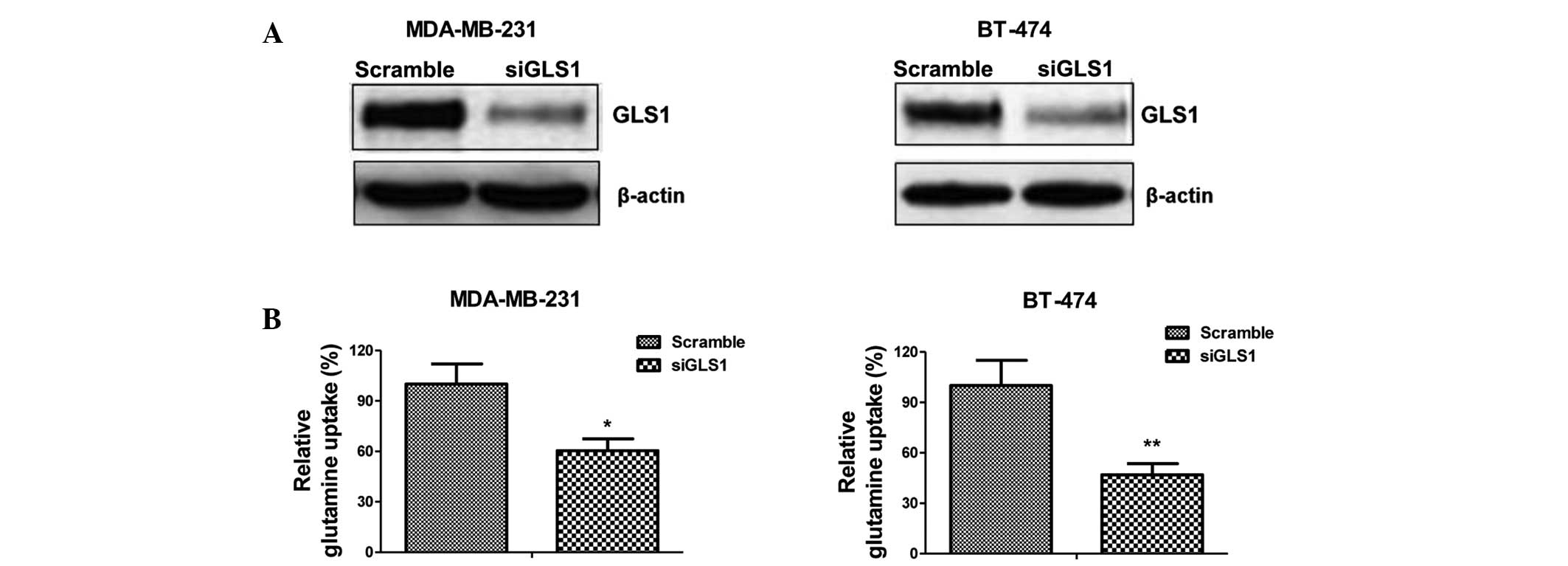
inhibition of glutamine metabolism. Knock down of GLS1 by siRNA
significantly inhibited the metabolism of glutamine in the
MDA-MB-231 and BT-474 cells (Fig.
4A and B). The glutamine uptake of the MDA-MD-231 and BT-474
cells decreased by 40 and 50%, respectively, compared with the
siRNA-transfected control (Fig.
4B). The present study subsequently examined whether GLS1
knockdown using siRNA in Taxol-resistant cells resulted in
re-sensitization of the cells to Taxol. The expression of GLS1 was
reduced using siRNA in the MDA-MB-231 parental cells and the
Taxol-resistant cells, followed by treatment with Taxol at
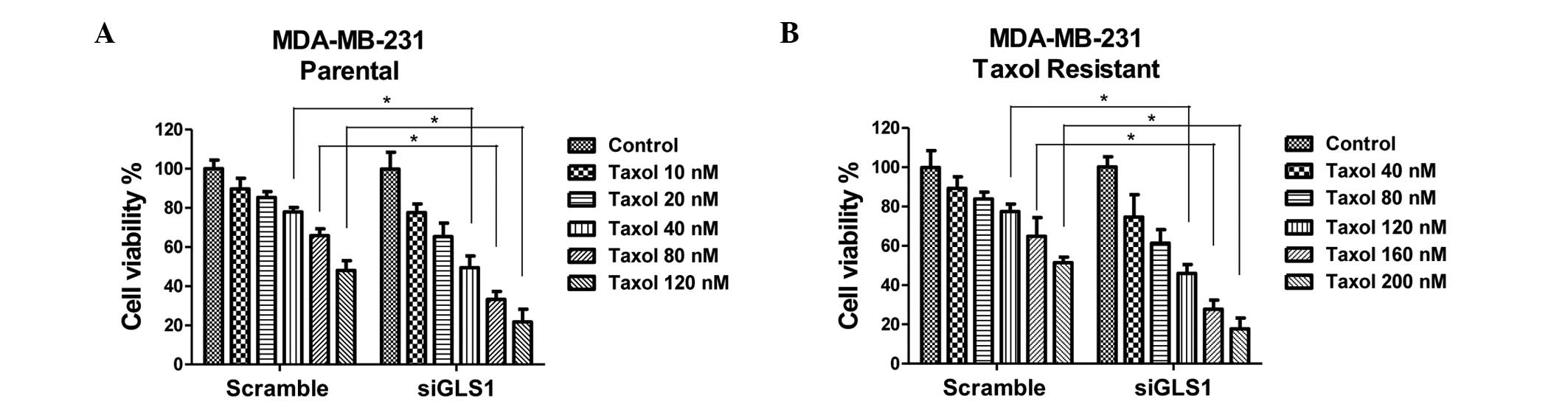
different concentrations for 48 h. The data revealed that
knock-down of GLS1 in the parental cells and the Taxol-resistant
cells rendered them sensitive to Taxol (Fig. 5). Cells with a lower expression of
GLS1 exhibited reduced viability following treatment with Taxol.
These results supported the hypothesis that inhibition of glutamine
metabolism by knocking down GLS1 resensitizes Taxol-resistant
cancer cells to Taxol.
Discussion
The ‘Warburg effect’ describes the characteristic of
cancer cells to produce energy predominantly from the glycolytic
breakdown of glucose, rather than mitochondrial oxidative
phosphorylation (13). In
addition, cancer cells exhibit other metabolic characteristics,
including increased fatty acid synthesis and glutamine metabolism
(13). Glutamine, the most
abundant amino acid in the blood, is important in cell growth and
metabolism. Cancer cells rely on glutamine metabolism for increased
production of by-products, which are necessary for rapidly
proliferating cells, including amino-acid precursors (16). Additionally, glutaminolysis
represents a fundamental mechanism for nitrogen anabolism (16,19).
It has been reported that glutaminolysis is
associated with drug resistance via the activation of mammalian
target of rapamycin complex 1 signaling in gastric cancer (20). The present study demonstrated that
glutamine metabolism was induced by treatment with Taxol in human
breast cancer cells. According to this phenotype, Taxol-resistant
cells were established from the MDA-MB-231 cell line, and the
results revealed that the Taxol-resistant cells exhibited
upregulated glutamine uptake rate compared with the parental cells.
This indicated that dysregulated glutamine metabolism may be a
target for the development of therapeutic drugs for overcoming
Taxol resistance in patients with cancer.
The first stage of glutamine metabolism involves GLS
catalyzing the conversion of glutamine to glutamate. The present
study revealed that the expression of GLS1 was upregulated in
Taxol-resistant cells. It has been reported that the overexpression
of GLS1 correlates with cell proliferation and tumor growth
(21). Inhibition of GLS prevents
oncogenic transformation and slows cell growth in certain types of
glioma (22). The present
investigation demonstrated that the overexpression of GLS1 rendered
breast cancer cells resistant to Taxol, suggesting a close
association between GLS1 and Taxol resistance. Inhibition of the
expression of GLS1 using siRNA significantly resensitized
Taxol-resistant cells to Taxol, indicating that, overexpression of
GLS1 may be the mechanism underlying Taxol resistance in cancer
cells. It is possible that tumor-initiating oncogenes may promote
glutamine utilization through elevating the expression of GLS as
part of the metabolic transformation process. Overexpressed
oncogenes, including c-Myc, have been demonstrated to increase the
expression of GLS1 in human tumor cells (23). In addition, ERBB2 has been
suggested to contribute to Taxol resistance by promoting glycolysis
in cancer cells (10). A previous
investigation reported that the activation of ERBB2 upregulates the
expression of GLS1, which promotes the proliferation of breast
cancer cells (24). This indicates
that upregulated expression levels of GLS1 and glutamine metabolism
may be the mechanism underlying ERBB2-induced Taxol resistance.
Further investigation aims to examine the detailed mechanism of
Taxol-induced glutamine metabolism in cancer cells and identify
inhibitors of glutamine metabolism for the development of
therapeutic strategies to overcoming drug resistance.
Acknowledgments
The authors would like to thank the staff and
faculty of the Department of Medical Oncology, Yantai Yantaishan
Hospital (Yhina, China), including Dr Yaobo Song for their
editorial assistance.
References
|
1
|
Henley D, Isbill M, Fernando R, Foster JS
and Wimalasena J: Paclitaxel induced apoptosis in breast cancer
cells requires cell cycle transit but not Cdc2 activity. Cancer
Chemother Pharmacol. 59:235–249. 2007. View Article : Google Scholar
|
|
2
|
Tan M and Yu D: Molecular mechanisms of
ERBB2-mediated breast cancer chemoresistance. Adv Exp Med Biol.
608:119–129. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Orr GA, Verdier-Pinard P, McDaid H and
Horwitz SB: Mechanisms of Taxol resistance related to microtubules.
Oncogene. 22:7280–7295. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Frankel A, Buckman R and Kerbel RS:
Abrogation of taxol-induced G2-M arrest and apoptosis in human
ovarian cancer cells grown as multicellular tumor spheroids. Cancer
Res. 57:2388–2293. 1997.PubMed/NCBI
|
|
5
|
Yin S, Bhattacharya R and Cabral F: Human
mutations that confer paclitaxel resistance. Mol Cancer Ther.
9:327–335. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zaffaroni N, Pennati M, Colella G, Perego
P, Supino R, Gatti L, Pilotti S, Zunino F and Daidone MG:
Expression of the anti-apoptotic gene survivin correlates with
taxol resistance in human ovarian cancer. Cell Mol Life Sci.
59:1406–1412. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wertz IE, Kusam S, Lam C, Okamoto T,
Sandoval W, Anderson DJ, Helgason E, Ernst JA, Eby M, Liu J,
Belmont LD, Kaminker JS, O’Rourke KM, Pujara K, Kohli PB, Johnson
AR, Chiu ML, Lill JR, Jackson PK, Fairbrother WJ, Seshagiri S,
Ludlam MJ, Leong KG, Dueber EC, Maecker H, Huang DC and Dixit VM:
Sensitivity to antitubulin chemotherapeutics is regulated by MCL1
and FBW7. Nature. 471:110–114. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ferlini C, Cicchillitti L, Raspaglio G,
Bartollino S, Cimitan S, Bertucci C, Mozzetti S, Gallo D, Persico
M, Fattorusso C, Campiani G and Scambia G: Paclitaxel directly
binds to Bcl-2 and functionally mimics activity of Nur77. Cancer
Res. 69:6906–6914. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yu D, Liu B, Jing T, Sun D, Price JE,
Singletary SE, Ibrahim N, Hortobagyi GN and Hung MC: Overexpression
of both p185c-ERBB2 and p170mdr-1 renders breast cancer cells
highly resistant to taxol. Oncogene. 16:2087–2094. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ding Y, Liu Z, Desai S, Zhao Y, Liu H,
Pannell LK, Yi H, Wright ER, Owen LB, Dean-Colomb W, Fodstad O, Lu
J, LeDoux SP, Wilson GL and Tan M: Receptor tyrosine kinase ERBB2
translocates into mitochondria and regulates cellular metabolism.
Nat Commun. 3:12712012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim SH, Juhnn YS and Song YS: Akt
involvement in paclitaxel chemoresistance of human ovarian cancer
cells. Ann NY Acad Sci. 1095:82–89. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen T, Pengetnze Y and Taylor CC: Src
inhibition enhances paclitaxel cytotoxicity in ovarian cancer cells
by caspase-9-independent activation of caspase-3. Mol Cancer Ther.
4:217–224. 2005.PubMed/NCBI
|
|
13
|
Zhao Y, Butler EB and Tan M: Targeting
cellular metabolism to improve cancer therapeutics. Cell Death Dis.
4:e5322013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Aledo JC, Gómez-Fabre PM, Olalla L and
Marquez J: Identification of two human glutaminase loci and
tissue-specific expression of the two related genes. Mamm Genome.
11:1107–1110. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gómez-Fabre PM, Aledo JC, Del
Castillo-Olivares A, Alonso FJ, Nunez De Castro I, Campos JA and
Marquez J: Molecular cloning, sequencing and expression studies of
the human breast cancer cell glutaminase. Biochem J. 345:365–375.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dang CV: Glutaminolysis: supplying carbon
or nitrogen or both for cancer cells? Cell Cycle. 9:3884–3886.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wise DR and Thompson CB: Glutamine
addiction: a new therapeutic target in cancer. Trends Biochem Sci.
35:427–433. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhou M, Zhao Y, Ding Y, Liu H, Liu Z,
Fodstad O, Riker AI, Kamarajugadda S, Lu J, Owen LB, Ledoux SP and
Tan M: Warburg effect in chemosensitivity: targeting lactate
dehydrogenase-A re-sensitizes taxol-resistant cancer cells to
taxol. Mol Cancer. 9:332010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Meng M, Chen S, Lao T, Liang D and Sang N:
Nitrogen anabolism underlies the importance of glutaminolysis in
proliferating cells. Cell Cycle. 9:3921–3932. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Durán RV, Oppliger W, Robitaille AM,
Heiserich L, Skendaj R, Gottlieb E, et al: Glutaminolysis activates
Rag-mTORC1 signaling. Mol Cell. 47:349–358. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
de la Rosa V, Campos-Sandoval JA,
Martin-Rufian M, Cardona C, Mates JM, Segura JA, Alonso FJ and
Marquez J: A novel glutaminase isoform in mammalian tissues.
Neurochem Int. 55:76–84. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Seltzer MJ, Bennett BD, Joshi AD, Gao P,
Thomas AG, Ferraris DV, Tsukamoto T, Rojas CJ, Slusher BS,
Rabinowitz JD, et al: Inhibition of glutaminase preferentially
slows growth of glioma cells with mutant IDH1. Cancer Res.
70:8981–8987. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gao P, Tchernyshyov I, Chang TC, Lee YS,
Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT and
Dang CV: c-Myc suppression of miR-23a/b enhances mitochondrial
glutaminase expression and glutamine metabolism. Nature.
458:762–765. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Qie S, Chu C, Li W, Wang C and Sang N:
ERBB2 activation upregulates glutaminase 1 expression which
promotes breast cancer cell proliferation. J Cell Biochem.
115:498–509. 2014. View Article : Google Scholar
|