Introduction
Human Burkitt’s Lymphoma (BL) originates from the
lymphatic system and was first described in children in central
Africa in 1956 (1).
Immunodeficiency, Epstein-Barr virus (EBV) infection and c-Myc gene
translocation have been associated with the development of this
malignant disease (2–4). In the human BL Raji cell line, EBV
DNA has been reported to integrate into the host genome, leading to
identification and isolation of this virus (5). Despite the marked potency of the
prevalent regimen of dose-adjusted treatment with etoposide,
prednisone, vincristine, cyclophosphamide, doxorubin and rituximab
in suppressing Burkitt’s lymphoma (6), the majority of patients succumb to
mortality within five years.
EBV is a ubiquitous human herpes-virus, which is
associated with the development of lymphoid and epithelial tumors
(3). The transforming effects of
EBV are dependent on its restricted expression of latent proteins
in the virally-infected epithelial cells and B-lymohoblastoid cell
lines (7,8). LMP1 is the major transforming protein
of EBV, which has been observed to act as a classical oncoprotein
in rodent fibroblast transforming assays, and is essential for
EBV-induced transformation (9).
Previously, EBV-induced oxidative stress in B lymphocytes and
epithelial cells has been reported to be involved in viral
transformation (10). In addition,
EBV-positive BL cells have been found to exhibit higher levels of
reactive oxygen species (ROS) compared with EBV-negative BL cells,
and latent membrane protein 1 (LMP1) is considered to be a major
inducer of ROS (11). As a
functional homologue of CD40, a member of the tumor necrosis factor
(TNF) receptor family, LMP1 can constitutively activate the TNF
pathway and, therefore, stimulate a number of downstream signaling
pathways, including nuclear factor (NF)-κB, mitogen-activated
protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K)/Akt
(12–15). Ha and Lee (16) demonstrated that the activation of
CD40 produces ROS by activating the NADPH oxidase (NOX) regulatory
subunit, p40phox, through TNF receptor-associated factor 3 and the
PI3K pathway. These findings imply that NOX may be involved in
LMP1-induced ROS induction in human malignancy, however, the
detailed molecular mechanism remains to be elucidated. The NOX
family is an important intrinsic source of ROS. By consuming NADPH,
NOX catalyzes the generation of superoxide from oxygen, and the
activation of NOX has been associated with development of various
types of tumor, including human prostate cancer (17,18),
melanoma (19) and glioblastoma
(20). In the EBV-induced
malignant transformation and tumor progression of B-cell lymphoma
cells, EBV nuclear antigen (EBNA)-1 has been suggested to activate
NOX2/gp91(phox) and facilitate ROS production (21).
ROS accumulation in malignant cells leads to their
increased sensitivity to ROS-induction anticancer compounds,
including β-phenylethyl isothiocyanate and cisplatin (22,23).
Our previous study demonstrated that the small molecular compound,
apogossypolone (ApoG2), not only inhibits the B cell lymphoma
(Bcl)-2 anti-apoptotic protein, but also induce ROS generation in
nasopharyngeal carcinoma cells (24,25).
The present study detected ROS levels, and NAPDH oxidase activity
in EBV-positive Raji cells and EBV-negative malignant B cells. In
addition, ROS accumulation, and cell cycle progression and
regulation were analyzed following treatment with ApoG2.
Materials and methods
Cell lines, chemicals and reagents
The EBV-positive Raji cells, a well-established
human BL cell line with EBV-latent infection; human pre-B leukemic
697 cells, human hematopoietic B NALM 6 cells and acute
lymphoblastic leukemia-derived REH cells were maintained in the Sun
Yat-sen University Cancer Center laboratory (Guangzhou, China) in
RPMI-1640 (Gibco-BRL, Gaithersburg, MD, USA), supplemented with 10%
heat-inactivated fetal bovine serum (Thermo Fihser Scientific,
HyClone, Logan, UT, USA). The cells were incubated in a humidified
5% CO2 atmosphere at 37°C. Apogossypolone (ApoG2) was
provided by Professor Dajun Yang (Ascenta Therapeutics, Inc.,
Malvern, PA, USA) and was dissolved in dimethyl sulfoxide (DMSO)
and freshly diluted in RPMI-1640 prior to used. The final DMSO
concentration was <0.1% (v/v). 2′,7′-dichlorodihydrofluorescein
diacetate (CM-H2DCF-DA) was purchased from Invitrogen Life
Technologies, Carlsbad, CA), USA.
Flow cytometry
Raji, REH, 697 and NALM16 cells (5×106)
were treated by 10 μM ApoG2 for 3 h, then the cellular ROS
contents were measured by incubating the drug-treated Raji, REH,
697 and NALM16 cells with 1 μM CM-H2DCF-DA for 60 min at
37°C, followed by flow cytometry, using a FACSCalibur equipped with
CellQuest Pro software version 3.3 (BD Biosciences, Franklin Lakes,
CA, USA). CM-H2DCF-DA is a fluorescent probe, which is relatively
specific for hydrogen peroxide (38). To determine the effect of ApoG2 on
cell cycle, Raji cells were treated with 10 μM ApoG2 for 8
and 16 h. Then untreated and drug-treated Raji cells
(5×106) were harvested. Propidium iodide (PI) staining
was performed following 75% alcohol fixation, which was then
analyzed using flow cytometry. The cellular DNA content was
determined using a flow cytometer (FC 500 MCL; Beckman Coulter,
Brea, CA, USA). Apoptotic cells were identified by the sub-G1 phase
in the cell cycle distribution.
NOX activity assay
DPI is widely used as an inhibitor of flavoenzymes,
particularly NADPH oxidases (19,20).
To determine the cellular NOX activity, 10 μM DPI
(Sigma-Aldrich, St. Louis, MO, USA) was added 4 h prior to
harvesting the cells. The control and DPI-treated Raji and NALM6,
697 and REH cells (5×106) were lysed in hypotonic
phosphate buffer containing protease inhibitors (Sigma-Aldrich),
and were then disrupted by sonication (Q125; Qsonica LLC, Newtown,
CT, USA), centrifuged for 10 min at 200 × g. The supernatant, which
contained the cytosol and mitochondrial fraction, was further
ultra-centrifuged at 100,000 × g for 30 min at 4°C. The resulting
pellet, containing the membranous fraction of cytosol and
mitochondria, was resuspended in buffer B, containing 50 mM Tris
(pH 7.5), 150 mM NaCl, 1 mM EDTA, protease inhibitor cocktail (one
tablet for 10 ml buffer; Thermo Fisher Scientific, Waltham, MA,
USA) for the NOX activity assay. The samples were adjusted to each
contain the same concentration of proteins (1 μg/μl),
and 5 μl of each sample was incubated with 94 μl
phosphate buffer, containing 50 mM K2HPO4, 1
mM EGTA, 150 mM sucrose, and 1 μl 3-NADPH (4 mM) for 15 min,
and 2.5 μl lucigenin (2 mM; Anaspec, Inc., Fremont, CA, USA)
was added prior to analysis. The lucigenin-derived
chemiluminescence of the cell homogenates were assessed over a
period of 1 min in a 20/20 n Tube Luminometer (Turner Biosystem,
Sunnyvale, CA).
Assays for cytotoxicity
The cell viability was measured using a
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethox
yphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (MTS) assay (cat. no.
CTB169; Promega Corporation, Madison, WI, USA). MTS, in the
presence of phenazien methosulfate, produces a formazan product,
which has an absorbance maximum of 490–500 nm in PBS and was
quantified using a spectrophotometer (V-530; JASCO, Tokyo, Japan).
The Raji cells were plated in 96-well culture clusters (Costar,
Cambridge, MA, USA) at a density of 10,000 cells/ml. Serial
dilutions were made from stock solution of ApoG2 to desired
concentrations (0.03, 0.3 and 3 μM). All the experimental
concentrations had three replicates. The percentage absorbance
relative to the control was plotted as a linear function of drug
concentration.
Western blot analysis
Protein analyses by immunoblotting were performed,
as previously described (39),
using primary antibodies against c-Myc (cat. no. sc-764; Santa Cruz
Biotechnology, Inc. Santa Cruz, CA, USA),
chromodomain-helicase-DNA-binding protein 1 (CHD1; cat. no. ab3242;
Abcam), checkpoint kinase 1 (Chk1; cat. no. 2345; Cell Signaling
Technology, Inc. Danvers, MA, USA), cell division cycle 25A
(cdc25A; cat. no. sc-97; Santa Cruz Biotechnology, Inc.), DNA
damage-binding protein 1 (DDB1; cat. no. 5428; Cell Signaling
Technology, Inc.), cyclin-dependent kinase 2 (CDK2) (cat. no.
sc-163; Santa Cruz Biotechnology, Inc.), cyclin E (cat. no. sc-481;
Santa Cruz, Inc.), cytochrome c (cat. no. 4272; Cell
Signaling Technology, Inc.) and B cell lymphoma-associated X
protein (Bax; cat. no. 2772; Cell Signaling Technology, Inc.). The
total cell lysates were harvested, electrophoresed by 12%
SDS-polyacrylamide gel (Sigma-Aldrich) electrophoresis and then
transferred onto polyvinylidene difluoride membranes (Roche, Basel,
Switzerland). The membranes were incubated with the antibodies
targeting c-Myc, CHD1, Chk1, cdc25A, CDK2, cyclin E, cytochrome
c and Bax (1:1,000 dilution in blocking buffer) overnight at
4°C with gentle agitation. Following incubation with the primary
antibodies, the membranes were washed in tris-buffered saline-Tween
20 and incubated with anti-rabbit (#7071) and anti-mouse (#7072)
horseradish peroxidase-conjugated secondary antibodies (Cell
Signaling Technology, Inc.; 1:2000 dilution in blocking buffer) for
1 h at room temperature, to facilitate detection, and enhanced
chemiluminescence reagent (Cell Signaling Technology, Inc.) to
develop the blots.
Detection of cell apoptosis
The ApoG2-induced apoptosis in the Raji cells was
evaluated using 4, 6-diamidine-2-phenylin-dole (DAPI; Beyotime,
Shanghai, China) nuclear staining. The cells were cultured in
6-well cell culture clusters at 37°C and exposed to 0, 5, 10 and 20
μM ApoG2 for 48 h. For fluorescent microscopic examination,
the cells were fixed with 10% absolute methanol, permeabilized
using 0.5% Triton-X-100, and stained with DAPI (2 μg/ml) for
30 min at 37°C. Subsequently, morphological changes of the nuclei
were examined, to assess for apoptotic characteristics by
fluorescence microscopy (DFC480; Leica Microsystems, Heidelberg
GmbH, Mannheim, Germany).
Statistical analysis
All statistical analyses, to determine the
significance of the data, were performed using one way analysis of
variance with SPSS 19.0 software (SPSS, Inc., St. Louis, MO, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
Excessive generation of ROS and NOX
activation in EBV-positive Raji lymphoma cells
Increased ROS generation is common in cancer cells
with an active glycolytic rate under the influence of oncogenic
signals, including EBV infection. It has been demonstrated that
EBNA-1 activates NOX2/gp91phox and is responsible for excessive ROS
production (21). This ROS
accumulation renders cells vulnerable to further oxidative damage
by exogenous agents. To investigate the role of EBV on cellular ROS
levels and NOX activation in lymphocytes, three EBV-negative
lymphocytes, 697, NALM 6 and REH, and EBV-positive Raji cells were
used. As shown in Fig. 1A, the
flow cytometric analysis indicated that the Raji BL cells exhibited
markedly higher basal ROS levels compared with the REH, 697 and
NALM6 cells (7-, 9.7- and 35.9-fold, respectively; Fig. 1A). Consistent with the this, the
NOX enzyme activity in the Raji cells was also ~2.0-, 2.5- and
5.5-fold higher compared with the NALM6, 697 and REH cells,
respectively (Fig. 1B). As EBNA1
initiates latent viral replication in B cells and regulates the
transcription of other EBV-encoded latent genes, this viral protein
is required for EBV latent infection and transformation in all EBV
associated tumors (26). A
previous study demonstrated that EBNA-1 is able to activate
NOX2/gp91(phox) in EBV-associated B-cell lymphoma cells (21). Based on these findings, the present
study hypothesized that EBV was able to induce excessive ROS
generation in EBV-related BL cells by activating NOX enzyme
activity. Excessive ROS levels in Raji BL cells facilitate cell
proliferation and tumorigenesis, making cancer cells exhibiting
high levels of ROS more sensitive to ROS-stimulatory anticancer
compounds (10). As shown in
Fig 1C, the small-molecular
compound, ApoG2, induced the generation of ROS >10-fold within 3
h following treatment in the Raji BL cells. This marked increase in
cellular ROS accumulation was hypothesized to result in severe
damage to cells.
 | Figure 1Activation of NOX in EBV-positive
Raji lymphoma cells. (A) Comparison of cellular ROS content in the
NALM6, 697, REH and Raji cells. Each histogram is representative of
three experiments (P<0.001, Raji cells, compared with. REH
cells). (B) Comparison of NOX activity in the NALM6, 697, REH and
Raji cells. Data are expressed as the mean ± standard deviation of
three experiments (*P<0.01 Raji cells, compared with
REH cells). (C) ApoG2 induced cellular ROS accumulation in the Raji
cells in 3 h. Each histogram is representative of three experiments
(P<0.001 compared with the untreated Raji cells). ApoG2,
apogossypolone; NOX, NADPH oxidase; CM-H2DCF-DA,
2′,7′-dichlorodihydrofluorescein diacetate. |
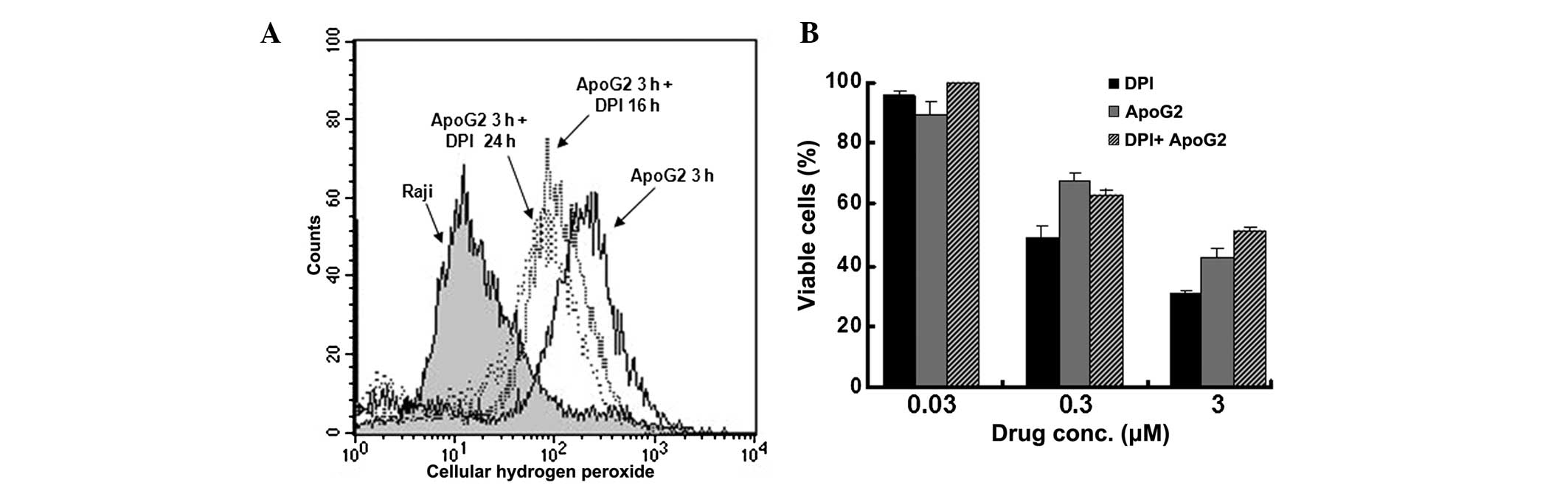
NOX inhibitor attenuates the
ApoG2-induced accumulation of ROS
To further assess the effect of cellular ROS levels
on cell proliferation in Raji cells, the cells were pretreated with
DPI, a specific inhibitor of the NOX enzyme, prior to treatment
with ApoG2. The flow cytometric analysis revealed that 10 μM
ApoG2 induced a significant increase in ROS (10.7-fold) in the Raji
cells, however, this increase was significantly lower following
pretreatment with DPI compared with the cells without DPI
pretreatment. ApoG2 induced lower levels of ROS in cells pretreated
with 5 μM DPI at 16 and 24 hr (6.2- and 4.0-fold,
respectively; Fig. 2A). DPI not
only attenuated the stimulatory effect of ApoG2 on ROS, but also
inhibited the antiproliferative activity of ApoG2 in the Raji
cells. As shown in Fig. 2B,
treatment with either DPI or ApoG2 alone exhibited dose-dependent
growth inhibition in the Raji cells following 72 h incubation,
however, when combined with DPI, ApoG2 inhibited growth less
compared with that observed without DPI. These results suggested
that, by inhibiting NOX, DPI attenuated the ROS-stimulatory effect
of ApoG2 and weakened the antiproliferative effect of ApoG2 in the
Raji BL cells.
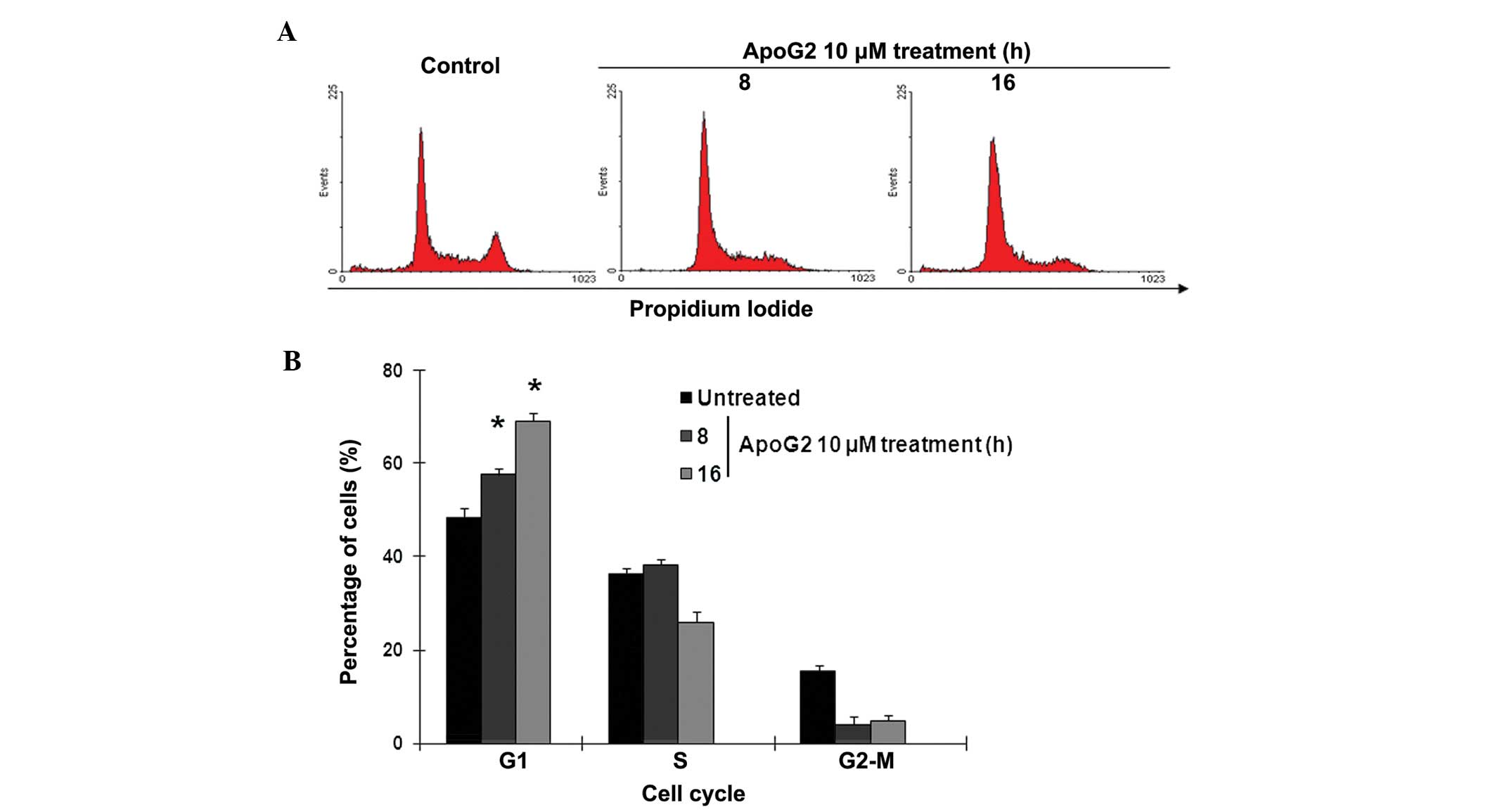
ApoG2 treatment arrests Raji cells in the
G1 phase of the cell cycle
Since ApoG2 treatment was observed to significantly
suppress cell proliferation (Fig.
2B), the present study investigated the effect of ApoG2-induced
ROS accumulation on cell proliferation in the Raji BL cells. To
examine the cell cycle proliferation, the cells were treated with
10 μM ApoG2 for 8 and 16 h. PI staining and flow cytometric
analysis demonstrated that ApoG2 was also unable to induce
apoptotic cell death within 16 h incubation, evidenced by low
sub-G1 signaling following PI-staining (Fig. 3A). Compared with 48% G1-cells in
the untreated control, almost 60 and 70% of the Raji cells were
arrested in the G1 phase of the cell cycle following treatment with
10 μM ApoG2 at 8 and 16 hr, respectively (Fig. 3A and B). These data, indicated that
ApoG2 stimulated ROS accumulation in the Raji BL cells within 3 h.
and this modification caused significant arrest at the G1 phase of
the cell cycle within 16 h. Although ApoG2 did not lead to cell
death in the short period of time following treatment, ApoG2
effectively suppressed cell proliferation, which was hypothesized
to cause significant cell death.
Cdh1, Chk1 and c-Myc cell cycle
regulators are involved in ApoG2-induced cell cycle arrest
To further investigate the molecular mechanism
underlying the ApoG2-induced cell cycle arrest, a panel of cell
cycle regulators were analyzed using immunoblotting. As shown in
Fig. 4A, the immunoblots
demonstrated that ApoG2 induced a significant downregulation of
c-Myc in the Raji cells, which was consistent with our previous
findings in NPC cells and U937 cells (27,28).
C-Myc appears to be sensitive to ROS stress, is non-specific
(29), and is not only affected by
a decrease in ROS, but also by ROS elevation, including ApoG2
treatment (25). In addition to
c-Myc, ApoG2 also suppressed the expression levels of Cdh1 and Chk1
as early as 3 and 6 h following treatment (Fig. 4B). However, ApoG2 did not cause
significant changes in the expression levels of cdc25, DDB1, cyclin
E and CDK2 in the Raji BL cells. Cdh1 has been reported to induce
G1/S arrest. These results suggested that the effects of ApoG2 on
cell cycle proliferation in Raji cells was likely mediated by a
pattern of modification of cell cycle regulators, including Cdh1,
Chk1 and c-Myc.
ApoG2 treatment induced apoptosis in Raji
BL cells
As the results of the PI staining and flow cytometry
indicated that ApoG2 did not induce cell death, but caused
significant cell cycle arrest within 16 h of treatment, the present
study examined weather the early damage resulted in cell death at a
later stage. To confirm this hypothesis, 5–20 μM ApoG2 was
used to treat cells for 48 h. As shown in Fig. 5A, PI staining and flow cytometric
analysis data indicated that, compared with the untreated control
cells, 10 and 20 μM ApoG2 induced a significant sub-G1 peak
(22 and 41%, respectively) in the Raji BL cells. DAPI staining
further demonstrated that 5 and 20 μM ApoG2 treatment for 48
h caused significant elevation in the number of apoptotic cells
with condensed chromatin (Fig. 5B and
C). To confirm the ApoG2-induced cell apoptosis in the Raji BL
cells, immunobloting was performed to examine the release of
cytochrome c and Bax from the mitochondria into cytosol. As
shown in Fig. 5D, the
immunobloting data demonstrated that cytochrome c was
significantly released into the cytosol by 5 μM ApoG2
treatment for 48 h. The levels of Bax in the cytosol also increased
in a dose-dependent manner 48 h after ApoG2 treatment. Taken
together, these findings suggested that ApoG2 was able to induce
significant apoptotic cell death a relatively long period of time
following treatment.
Discussion
Burkitt’s lymphoma (BL) is an aggressive malignant
disease with high morbidity and low survival rates. The incidence
of this disease has increased significantly worldwide as its
development is associated with viral infection and immunodeficiency
(30). EBV is a type of human
herpes virus, and is associated with the pathogenesis of BL. As
with several other oncoviruses, including hepatitis B virus and
human T-lymphotropic virus 1, which induce oxidative stress and
metabolic disorder in host cells (31,32),
EBV has also been found to upregulate cellular ROS levels through
the EBNA1-NOX signaling pathway (21). Consistent with these reports, the
present study found that, compared with the NALM6, 697 and REH
EBV-negative lymphocytes, the EBV-positive Raji cells exhibited
significantly higher levels of NOX enzyme activity and cellular
ROS. Based on these findings, it was suggested that NOX enzyme
activation and elevated ROS levels may be potent targets for
anticancer treatment in Raji BL cells or cancer cells with similar
characters.
It is understood that cancer cells with higher ROS
levels are more sensitive to ROS-stimulatory compounds, including
cisplatin and paclitaxel (22–23).
In the present study, ApoG2 demonstrated potent effects on ROS
stimulation as early as 3 h after treatment. This significant
increase in ROS content is likely to damage cell components,
including mitochondria and the nuclear and cell membranes. The
present study found that, as early as 8 h after ApoG2 treatment,
almost 60% of the Raji cells accumulated in the G1 phase of the
cell cycle. At 16 h after treatment, this percentage increased to
70%, although few cells had undergone apoptotic cell death,
evidenced by low sub-G1 signal in cells with PI staining. However,
a longer treatment duration of 48 h resulted in a marked sub-G1
peak and apoptotic cell death. These data suggested that the
ApoG2-induced ROS accumulation led to cell cycle arrest in the G1
phase at the early stage and caused apoptotic cell death at the
late stage.
Cell cycle progression is regulated by a panel of
molecules, including CDKs, cyclins, their regulators (33). Cdh1 is an adapter protein of
anaphase-promoting complex (APC) and controls the G0 and G1 phases
of the cell cycle. It stabilizes the G1 phase by recognizing
mitotic proteins, including cyclins, and recruits them to the APC
for ubiquination (34). In
neurons, actively upregulated APC/Cdh1 can constantly degrade
glycolysis enzyme 6-phosphofructo-2-kinase/fructose-2,
6-bisphosphatase-3 to support aerobic respiration and maintain
antioxidant status (35). A
previous study also reported that antioxidant treatment arrests
cells in the late-G1 phase by upregulating APC/Cdh1 activity
(36). Unlike the activation of
APC/Cdh1 by antioxidants, the present study demonstrated that the
ApoG2-induced ROS accumulation in the Raji BL cells suppressed the
expression of Cdh1 and arrested cells in the G1 phase. Chk1 was
also significantly suppressed by ApoG2 treatment. Chk1 is a kinase,
which phosphorylates cdc25 at Ser 216. Cdc25 is phosphorylated
during interphase, but not in mitosis. Phosphorylated cdc25 binds
to members of the 14–3–3 family of proteins, which prevents the
activation of cdc2 and prevents cells entering the M phase of the
cell cycle (33–34). The exposure of cells to excessive
ROS has been found to deplete cyclin D1 and activate Chk1 (37). In the present study, the protein
expression of Chk1 decreased as early as 6 h after ApoG2
treatment.
The c-Myc cell cycle regulator was also affected by
ApoG2 treatment in the present study. Our previous study
demonstrated that ApoG2 suppressed the levels of c-Myc in NPC cells
and U937 lymphoma cells (27,28).
In the present study c-Myc was downregulated by ApoG2 treatment in
the Raji BL cells. In addition, the NOX inhibitor, DPI, also
suppressed the protein expression of c-Myc within 12 h of treatment
(data not shown), which suggested that the protein suppression of
c-Myc was not specific to ApoG2, but was affected by ROS
accumulation and depletion.
In conclusion, the results of the present study
demonstrated that the EBV-positive Raji BL cells exhibited
increased cellular ROS content compared with the EBV-negative
lymphocytes, and this ROS elevation facilitated the sensitivity of
the Raji cells to the ROS-stimulatory compound, ApoG2. ApoG2
induced a significant increase in the levels of cellular ROS in the
Raji cells within 3 h of treatment. Within 8 h of treatment, ApoG2
arrested almost 60% of the Raji cells in the G1 phase of the cell
cycle, and the Cdh1 and Chk1 regulators may have been involved.
Within 48 h of treatment, ApoG2 induced significant apoptotic cell
death in the Raji BL cells. These findings indicate ApoG2 as a
promising compound for anti-BL treatment.
References
|
1
|
Davies JN, Elmes S, Hutt MS, Mtimavalye
LA, Owor R and Shaper L: Cancer in an African community, 1897 –
1956. An analysis of the records of Mengo hospital, Kampala, Uganda
2. Br Med J. 1:336–341. 1964. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Molyneux EM, Rochford R, Griffin B, et al:
Burkitt’s lymphoma. Lancet. 379:1234–1244. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bornkamm GW: Epstein-Barr virus and the
pathogenesis of Burkitt’s lymphoma: more questions than answers.
Int J Cancer. 124:1745–1755. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
zur Stadt U, Hoser G, Reiter A, Welte K
and Sykora KW: Application of long PCR to detect t(8;14)(q24;q32)
translocations in childhood Burkitt’s lymphoma and B-ALL. Ann
Oncol. 8(Suppl 1): 31–35. 1997. View Article : Google Scholar
|
|
5
|
Ansari MA, Singh VV, Dutta S, et al:
Constitutive interferon-inducible protein 16-inflammasome
activation during Epstein-Barr virus latency I, II, and III in B
and epithelial cells. J Virol. 87:8606–8623. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dunleavy K, Pittaluga S, Shovlin M, et al:
Low-intensity therapy in adults with Burkitt’s lymphoma. N Engl J
Med. 369:1915–1925. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Young LS and Murray PG: Epstein-Barr virus
and oncogenesis: from latent genes to tumours. Oncogene.
22:5108–5121. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chou YC, Lin SJ, Lu J, et al: Requirement
for LMP1-induced RON receptor tyrosine kinase in Epstein-Barr
virus-mediated B-cell proliferation. Blood. 118:1340–1349. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dawson CW, Port RJ and Young LS: The role
of the EBV-encoded latent membrane proteins LMP1 and LMP2 in the
pathogenesis of nasopharyngeal carcinoma (NPC). Semin Cancer Biol.
22:144–153. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lassoued S, Ben Ameur R, Ayadi W, Gargouri
B, Ben Mansour R and Attia H: Epstein-Barr virus induces an
oxidative stress during the early stages of infection in B
lymphocytes, epithelial, and lymphoblastoid cell lines. Mol Cell
Biochem. 313:179–186. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cerimele F, Battle T, Lynch R, et al:
Reactive oxygen signaling and MAPK activation distinguish
Epstein-Barr Virus (EBV)-positive versus EBV-negative Burkitt’s
lymphoma. Proc Natl Acad Sci USA. 102:175–179. 2005. View Article : Google Scholar
|
|
12
|
Kraus ZJ, Nakano H and Bishop GA: TRAF5 is
a critical mediator of in vitro signals and in vivo functions of
LMP1, the viral oncogenic mimic of CD40. Proc Natl Acad Sci USA.
106:17140–17145. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gewurz BE, Mar JC, Padi M, et al:
Canonical NF-kappaB activation is essential for Epstein-Barr virus
latent membrane protein 1 TES2/CTAR2 gene regulation. J Virol.
85:6764–6773. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shkoda A, Town JA, Griese J, et al: The
germinal center kinase TNIK is required for canonical NF-κB and JNK
signaling in B-cells by the EBV oncoprotein LMP1 and the CD40
receptor. PLoS Biol. 10:e10013762012. View Article : Google Scholar
|
|
15
|
Shair KH, Bendt KM, Edwards RH, Bedford
EC, Nielsen JN and Raab-Traub N: EBV latent membrane protein 1
activates Akt, NFkappaB, and Stat3 in B cell lymphomas. PLoS
Pathog. 3:e1662007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ha YJ and Lee JR: Role of TNF
receptor-associated factor 3 in the CD40 signaling by production of
reactive oxygen species through association with p40phox, a
cytosolic subunit of nicotinamide adenine dinucleotide phosphate
oxidase. J Immunol. 172:231–239. 2004. View Article : Google Scholar
|
|
17
|
Lim SD, Sun C, Lambeth JD, et al:
Increased Nox1 and hydrogen peroxide in prostate cancer. Prostate.
62:200–207. 2005. View Article : Google Scholar
|
|
18
|
Huang WC, Li X, Liu J, Lin J and Chung LW:
Activation of androgen receptor, lipogenesis, and oxidative stress
converged by SREBP-1 is responsible for regulating growth and
progression of prostate cancer cells. Mol Cancer Res. 10:133–142.
2012. View Article : Google Scholar :
|
|
19
|
Yamaura M, Mitsushita J, Furuta S, et al:
NADPH oxidase 4 contributes to transformation phenotype of melanoma
cells by regulating G2-M cell cycle progression. Cancer Res.
69:2647–2654. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hsieh CH, Shyu WC, Chiang CY, Kuo JW, Shen
WC and Liu RS: NADPH oxidase subunit 4-mediated reactive oxygen
species contribute to cycling hypoxia-promoted tumor progression in
glioblastoma multiforme. PLoS One. 6:e239452011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gruhne B, Sompallae R, Marescotti D,
Kamranvar SA, Gastaldello S and Masucci MG: The Epstein-Barr virus
nuclear antigen-1 promotes genomic instability via induction of
reactive oxygen species. Proc Natl Acad Sci USA. 106:2313–2318.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Trachootham D, Zhou Y, Zhang H, et al:
Selective killing of oncogenically transformed cells through a
ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer
Cell. 10:241–252. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Marullo R, Werner E, Degtyareva N, et al:
Cisplatin induces a mitochondrial-ROS response that contributes to
cytotoxicity depending on mitochondrial redox status and
bioenergetic functions. PLoS One. 8:e811622013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hu ZY, Zhu XF, Zhong ZD, et al: ApoG2, a
novel inhibitor of antiapoptotic Bcl-2 family proteins, induces
apoptosis and suppresses tumor growth in nasopharyngeal carcinoma
xeno-grafts. Int J Cancer. 123:2418–2429. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hu ZY, Wang J, Cheng G, et al:
Apogossypolone targets mitochondria and light enhances its
anticancer activity by stimulating generation of singlet oxygen and
reactive oxygen species. Chin J Cancer. 30:41–53. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Canaan A, Haviv I, Urban AE, et al: EBNA1
regulates cellular gene expression by binding cellular promoters.
Proc Natl Acad Sci USA. 106:22421–22426. 2009. View Article : Google Scholar
|
|
27
|
Hu ZY, Sun J, Zhu XF, Yang D and Zeng YX:
ApoG2 induces cell cycle arrest of nasopharyngeal carcinoma cells
by suppressing the c-Myc signaling pathway. J Transl Med. 7:742009.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sun J, Li ZM, Hu ZY, Zeng ZL, Yang DJ and
Jiang WQ: Apogossypolone inhibits cell growth by inducing cell
cycle arrest in U937 cells. Oncol Rep. 22:193–198. 2009.PubMed/NCBI
|
|
29
|
Maciag AE, Holland RJ, Robert Cheng YS, et
al: Nitric oxide-releasing prodrug triggers cancer cell death
through deregulation of cellular redox balance. Redox Biol.
1:115–124. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mbulaiteye SM, Anderson WF, Ferlay J, et
al: Pediatric, elderly, and emerging adult-onset peaks in Burkitt’s
lymphoma incidence diagnosed in four continents, excluding Africa.
Am J Hematol. 87:573–578. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gu JM, Lim SO, Oh SJ, Yoon SM, Seong JK
and Jung G: HBx modulates iron regulatory protein 1-mediated iron
metabolism via reactive oxygen species. Virus Res. 133:167–177.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Silic-Benussi M, Cavallari I, Vajente N,
et al: Redox regulation of T-cell turnover by the p13 protein of
human T-cell leukemia virus type 1: distinct effects in primary
versus transformed cells. Blood. 116:54–62. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ishidate T, Elewa A, Kim S, Mello CC and
Shirayama M: Divide and differentiate: CDK/Cyclins and the art of
development. Cell Cycle. 13:1384–1391. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li M, Shin YH, Hou L, et al: The adaptor
protein of the anaphase promoting complex Cdh1 is essential in
maintaining replicative lifespan and in learning and memory. Nat
Cell Biol. 10:1083–1089. 2008. View
Article : Google Scholar
|
|
35
|
Herrero-Mendez A, Almeida A, Fernández E,
Maestre C, Moncada S and Bolaños JP: The bioenergetic and
antioxidant status of neurons is controlled by continuous
degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell
Biol. 11:747–752. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Havens CG, Ho A, Yoshioka N and Dowdy SF:
Regulation of late G1/S phase transition and APC Cdh1 by reactive
oxygen species. Mol Cell Biol. 26:4701–4711. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pyo CW, Choi JH, Oh SM and Choi SY:
Oxidative stress-induced cyclin D1 depletion and its role in cell
cycle processing. Biochim Biophys Acta. 1830:5316–5325. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Pelicano H, Feng L, Zhou Y, et al:
Inhibition of mitochondrial respiration: a novel strategy to
enhance drug-induced apoptosis in human leukemia cells by a
reactive oxygen species-mediated mechanism. J Biol Chem.
278:37832–37839. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Guo C, Pan ZG, Li DJ, et al: The
expression of p63 is associated with the differential stage in
nasopharyngeal carcinoma and EBV infection. J Transl Med. 4:232006.
View Article : Google Scholar : PubMed/NCBI
|