Introduction
In recent decades, the incidence and prevalence of
type 2 diabetes (T2D) has significantly increased worldwide,
particularly among those aged between 35 and 40 years old (1). Long-term diabetes frequently induces
vascular changes and dysfunction, and diabetic vascular
complications are a major cause of morbidity and mortality amongst
patients with diabetes (2).
Dysfunction of the β cells of the Islets of
Langerhans and insulin-resistance are the main factors underlying
the development of T2D (3). In
addition, other factors, including inheritance, ethnicity and age,
are all associated with the incidence of T2D (4). A previous study identified that the
metabolism of glucose and fatty acids by skeletal muscle is
impaired in T2D (5). Reduced
glucose transport and phosphorylation, as well as reduced rates of
glycogen synthesis are common manifestations of insulin-resistant
glucose metabolism (6). However,
abnormal fatty acid metabolism involves the increased accumulation
of triglycerides and other lipids (7), as well as the dysregulation of lipid
oxidation under fasting and insulin-stimulated conditions (8). Consequently, the dysfunction of
metabolism in muscles may potentially indicate T2D.
DNA methylation is a key epigenetic modification of
the genome, which is involved in numerous cellular processes,
including cell differentiation and metabolism (9,10).
Consistent with these significant roles, associations between
aberrant DNA methylation and a growing number of human diseases
have been identified (11). In
addition, abnormal methylation of several genes, including
cytochrome c oxidase polypeptide 7A1, nicotinamide adenine
dinucleotide dehydrogenase (ubiquinone) 1 β subcomplex subunit 6,
peroxisome proliferator-activated receptor γ coactivator (PGC) 1-α
and PGC-1β, have been observed in tissues from the skeletal muscles
of individuals with type 2 diabetes (12–15).
The development of methylation chips has provided a
more efficient approach for the detection of global changes in DNA
methylation. Consequently, the current study utilized microarray
data derived from the muscle samples of eleven monozygotic twin
pairs, discordant for T2D, to detect the global changes in DNA
methylation of gene promoters associated with T2D. Gene Ontology
(GO) enrichment analysis and prediction analysis of binding sites
for potential transcription factors (TFs) were performed in order
to investigate the molecular mechanisms involved in T2D.
Materials and methods
Data acquisition
The DNA methylation dataset, GSE38291 (16), comprising data regarding 11 pairs
of monozygotic twins discordant for type 2 diabetes, was downloaded
from the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) database. Each pair
of monozygotic twins included one individual with T2D and one
normal individual (GSM938065-GSM938086). The Illumina
HumanMethylation27 BeadChips (Illumina Inc., San Diego, CA, USA),
containing a pair of methylated and unmethylated probes designed
for each CpG site, were used to detect the DNA methylation in the
gene promoter region.
Quantification of DNA methylation
signals
The raw intensities of methylated and unmethylated
probes for each sample were extracted based on the Illumina
HumanMethylation27 BeadChip. The intensity data were then
normalized using quantile normalization. The M-value of each CpG
site was calculated as the log2 ratio of the normalized
intensities of methylated probe versus unmethylated probe (17). The M-values of all probes in the
promoter of a single gene were calculated and the mid-value was
considered to be the methylation level of this gene promoter.
Analysis of differentially methylated
genes
A paired samples t-test was employed to analyze the
differentially methylated levels of gene promoter between T2D and
normal muscle samples. P<0.05 and a fold change of >1.3 were
considered to indicate the cutoff criteria.
GO enrichment analysis
GO enrichment analysis was performed on the
differentially methylated genes for biological processes and
cellular components using the online bioinformatics resource: The
Database for Annotation, Visualization and Integrated Discovery
version 6.7 (http://david.abcc.ncifcrf.gov/) (18). GO terms where P<0.1, were
considered to indicate significantly enriched GO terms.
Screening for potential TFs
The Whole-Genome rVISTA tool (19) was used to analyze the enriched TF
binding sites in the upstream region (200 bp, 500 bp and 1 kb) of
transcription start sites (TSS) of the abnormal hyper- and
hypo-methylated genes. The TFs with a P<0.01 were considered to
be significant. Subsequently, the significant TFs obtained from the
three regions were compared, and the most common TFs were
considered as the final TFs of the target genes.
Results
Differentially methylated genes
A total of 38 genes were identified to possess
differentially methylated regions between T2D and normal
monozygotic twins. Specifically, 22 genes, including SIRT1
(Sirtuin1), NAT6 (N-acetyltransferase 6) and PLA2G12B
(phospholipase A2 group XIIB), were hypermethylated in the T2D
group; while 16 genes, including nuclear factor of activated T
cells calcineurin-dependent 1 (NFATC1) and microtubule associated
serine/threonine kinase 1, were hypo-methylated (Table I, Fig.
1). In addition, two TFs were identified in the hyper- and
hypo- methylated genes, respectively, based on the public TF
database (Table I) (20). These two TFs were Runt-related
transcription factor 1 and NFATC1, which possessed significant
hyper- and hypo-methylated regions in T2D muscles.
 | Table ISignificant hyper- and hypo-methylated
genes in T2D. |
Table I
Significant hyper- and hypo-methylated
genes in T2D.
| Gene |
Log2FC | P-value | Transcription
factor |
|---|
| Hypermethylation in
T2D |
| GPATC1 | 0.770 | 0.006 | No |
| SMCX | 0.610 | 0.021 | No |
| MUC13 | 0.588 | 0.002 | No |
| PMVK | 0.569 | 0.042 | No |
| RPL18A | 0.503 | 0.019 | No |
| IL20RA | 0.486 | 0.025 | No |
| NAT6 | 0.481 | 0.006 | No |
| OIT3 | 0.472 | 0.011 | No |
| ARHGAP27 | 0.469 | 0.009 | No |
| RUNX1 | 0.468 | 0.015 | Yes |
| PFS2 | 0.466 | 0.038 | No |
| BRUNOL5 | 0.462 | 0.005 | No |
| CIAPIN1 | 0.443 | 0.012 | No |
| SPCS2 | 0.436 | 0.016 | No |
| MICB | 0.432 | 0.022 | No |
| MGAT3 | 0.431 | 0.039 | No |
| HSPA14 | 0.430 | 0.024 | No |
| LOC159090 | 0.426 | 0.049 | No |
| SIRT1 | 0.419 | 0.011 | No |
| C22orf9 | 0.413 | 0.039 | No |
| PLA2G12B | 0.409 | 0.029 | No |
| PPWD1 | 0.401 | 0.047 | No |
| Hypomethylation in
T2D |
| RECQL4 | −0.404 | 0.037 | No |
| SYT12 | −0.405 | 0.030 | No |
| GTF3C1 | −0.405 | 0.043 | No |
| NT5DC3 | −0.406 | 0.039 | No |
| NFATC1 | −0.406 | 0.002 | Yes |
| SETMAR | −0.412 | 0.025 | No |
| NOL7 | −0.421 | 0.031 | No |
| BICD2 | −0.424 | 0.032 | No |
| TCL6 | −0.430 | 0.031 | No |
| KCNE4 | −0.431 | 0.045 | No |
| DDOST | −0.448 | 0.004 | No |
| MAST1 | −0.450 | 0.005 | No |
| TYR | −0.451 | 0.020 | No |
| PURA | −0.468 | 0.038 | No |
| GNPDA1 | −0.471 | 0.022 | No |
| NEDD1 | −0.472 | 0.037 | No |
GO enrichment analysis
According to the GO enrichment analysis,
hyper-methylated genes were enriched in three GO terms (P<0.1,
Table II). The negative
regulation of response to stimulus (GO: 0048585), which was
associated with a process that stops, prevents or reduces the
frequency, rate or extent of a response to a stimulus, was enriched
by three genes, which may be significant in T2D pathology. The
terms cell death (GO: 0008219) and death (GO: 0016265), refer to
the biological processes that result in permanent cessation of all
vital functions of a cell. By contrast, the hypomethylated genes
were significantly enriched in three classes of GO terms. DNA
geometric change (GO: 0032392) is a term regarding the process in
which a transformation is induced in the geometry of a DNA double
helix, resulting in a change in twist and/or writhe. There were
three genes enriched for DNA metabolic process (GO: 0006259), which
is associated with cellular metabolic processes involving
deoxyribonucleic acid. Finally, two genes were highly enriched for
DNA recombination (GO: 0006310), involving the process in which a
new genotype is formed via a reassortment of genes resulting in
novel gene combinations.
| Table IISignificantly enriched GO terms of
differentially methylated genes in T2D. |
Table II
Significantly enriched GO terms of
differentially methylated genes in T2D.
| Methylation
status | GO term | Gene counts | P-value | Genes |
|---|
| Hypermethylated
genes in T2D | GO:
0048585-negative regulation of response to stimulus | 2 | 0.072 | MICB, SIRT1 |
| GO: 0008219-cell
death | 3 | 0.096 | MICB, CIAPIN1,
SIRT1 |
| GO:
0016265-death | 3 | 0.097 | MICB, CIAPIN1,
SIRT1 |
| Hypomethylated
genes in T2D | GO: 0032392-DNA
geometric change | 2 | 0.015 | RECQL4, PURA |
| GO: 0006259-DNA
metabolic process | 3 | 0.061 | RECQL4, SETMAR,
PURA |
| GO: 0006310-DNA
recombination | 2 | 0.082 | RECQL4, SETMAR |
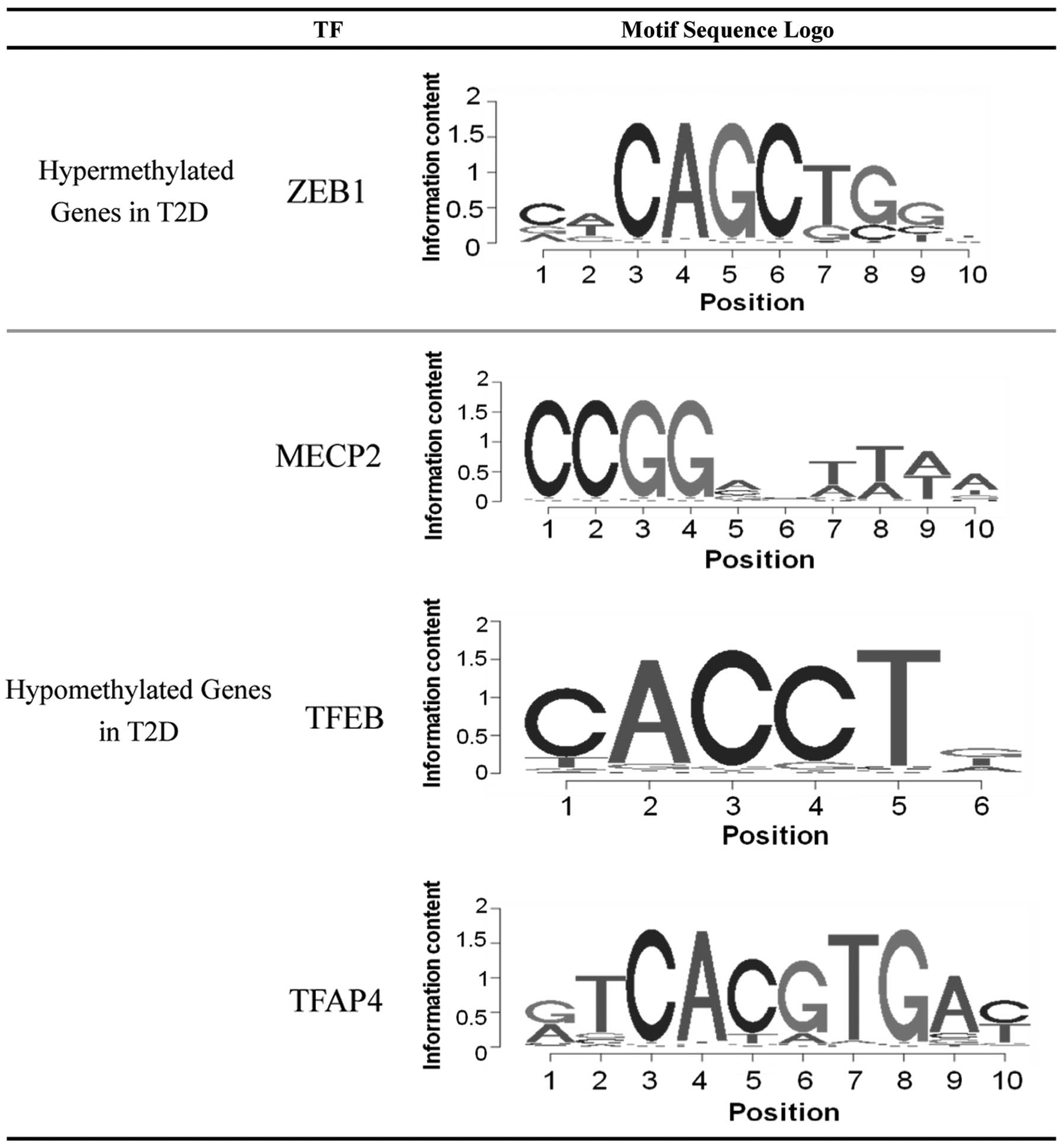
Screening for potential TFs
The enrichment of TF binding sites in the upstream
region of the abnormal hyper- and hypomethylated genes was
analyzed. Multiple TF binding sites were significantly enriched
among the differentially methylated genes, and four corresponding
TFs were subsequently identified. As shown in Fig. 2, only the TF zinc finger E-box
binding homeobox 1 (ZEB1) was identified as possessing a large
probability to bind and regulate the hyper-methylated genes. While
the binding sites of three TFs, methyl CpG binding protein 2
(MECP2), TFEB and TFAP4, were enriched in the hypo-methylated
genes.
Discussion
The differences in global DNA methylation in muscle
tissues from 11 monozygotic twin pairs discordant for T2D were
detected using microarray data. The utilization of mono-zygotic
twins facilitates investigation of the abnormal DNA methylation
induced by environmental factors, but not hereditary factors, in
T2D patients.
The significantly hyper- and hypo-methylated genes
identified in the current study may provide several novel candidate
genes associated with the occurrence of T2D. SIRT1, an
NAD(+)-dependent histone deacetylase, which was hyper-methylated
according to the present results, was observed to regulate
glucose/lipid metabolism through its deacetylase activity on
numerous substrates (21,22). In addition, SIRT1 positively
regulates insulin secretion in pancreatic β cells (23), and protects cells from oxidative
stress and inflammation. Consequently, the methylation of the SIRT1
gene may be a significant gene modification involved in the
occurrence of T2D. The other two genes, NAT6 and PLA2G12B, which
were also hyper-methylated in T2D muscle, are involved in the
metabolism of lipids (24).
PLA2G12B belongs to the PLA2 family, and catalyzes the hydrolysis
of glycolipids to release free fatty acids and lysophospholipids
(25). NAT6 encodes a cytoplasmic
N-acetyltransferase protein, with substrate specificity for
proteins with an N-terminal methionine (26). Additionally, these two genes are
involved in the pathway of glycerophospholipid metabolism according
to the Kyoto Encyclopedia of Genes and Genomes pathway analysis
(http://www.genome.jp/kegg/). All of this
cumulative evidence has suggested that the hyper-methylation of
PLA2G12B and NAT6 may result in the dysfunction of
glycerophospholipid metabolism leading to hyperlipidemia in T2D
patients.
Of the hypo-methylated genes, NFATC1 is particularly
notable for its potential association with T2D. A previous study
revealed that mice with a β-cell-specific deletion of the
calci-neurin b1 (CNB1) subunit developed age-dependent diabetes
characterized by decreased β-cell proliferation and mass, reduced
pancreatic insulin content and hypo-insulinemia. The expression of
active NFATC1 in CNB1-deficient β cells may rescue these defects
and prevent diabetes (27). This
evidence indicated the positive role of NFATC1 in the pathogenesis
or the potential therapeutic role in diabetes.
According to the enrichment of TF binding sites in
the upstream region of the differentially methylated genes, it was
demonstrated that the transcription factor ZEB1 was significantly
enriched for its binding to the hyper-methylated genes identified
in the current study. Papadopoulou et al (28) hypothesized that transfection of
ZEB1 cDNA may induce a reduction in B cell lymphoma 6 (BCL6)
expression, and that siRNA-mediated knockdown of ZEB1 may lead to
an increase in BCL6 mRNA expression (28). BCL-6 represses menin, a product of
the Men1 gene, expression. Transgenic menin overexpression in β
cells prevents islet expansion, which leads to maternal
hyperglycemia (29). Therefore,
transgenic menin mice exhibit features of human gestational
diabetes (29). For these reasons,
ZEB1, as a transcription factor, may be involved in the
pathogenesis of T2D. The binding site of MECP2 was significantly
enriched by the hypo-methylated genes in the present study. MECP2
is capable of binding specifically to methylated DNA to suppress
transcription from methylated gene promoters (30). The DNA methylation of genes, as the
major modification of eukaryotic genomes (31), was repressed by MECP2, indicating a
key function for MECP2 in the regulation of methylated genes. It
was therefore hypothesized that MECP2 may be involved in the
process of T2D via regulation of the hypo-methylated genes;
however, this hypothesis requires further investigation for
confirmation. The present results also identified two candidate TFs
(TFEB and TFAP4), which may regulate the hypo-methylated genes in
T2D. To the best of our knowledge, few studies have investigated
the association between these two TFs and T2D, and therefore future
studies should focus on elucidating this association.
In conclusion, a bioinformatics analysis of the
differentially methylated genes in muscle tissues from 11
monozygotic twin pairs discordant for T2D was performed. The
identified diabetes-associated genes (SIRT1, NAT6, PLA2G12B and
NFATC1) and potential TFs provided a novel insight into the
molecular mechanism underlying the occurrence of T2D. These genes
may become promising target genes in the development of novel
treatments for T2D.
Acknowledgments
The present study was supported by a grant from a
project supported by the China National Natural Science Foundation
(grant no. 81300260) and the Foundation of Science and Technology
Department of Sichuan Province (grant no. 14ZC1146).
References
|
1
|
Parving H-H, Brenner BM, McMurray JJ, et
al ALTITUDE Investigators: Cardiorenal end points in a trial of
aliskiren for type 2 diabetes. N Engl J Med. 367:2204–2213. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kitada M and Koya D: SIRT1 in type 2
diabetes: Mechanisms and therapeutic potential. Diabetes Metab J.
37:315–325. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kahn SE: The relative contributions of
insulin resistance and beta-cell dysfunction to the pathophysiology
of Type 2 diabetes. Diabetologia. 46:3–19. 2003.PubMed/NCBI
|
|
4
|
Spranger J, Kroke A, Möhlig M, et al:
Adiponectin and protection against type 2 diabetes mellitus.
Lancet. 361:226–228. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hofsø D, Jenssen T, Hager H, Røislien J
and Hjelmesaeth J: Fasting plasma glucose in the screening for type
2 diabetes in morbidly obese subjects. Obes Surg. 20:302–307. 2010.
View Article : Google Scholar
|
|
6
|
Zhang P, Zhang X, Brown J, et al: Global
healthcare expenditure on diabetes for 2010 and 2030. Diabetes Res
Clin Pract. 87:293–301. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liebl A, Mata M and Eschwège E; ODE-2
Advisory Board: Evaluation of risk factors for development of
complications in Type II diabetes in Europe. Diabetologia.
45:S23–S28. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sjöström L, Lindroos A-K, Peltonen M, et
al Swedish Obese Subjects Study Scientific Group: Lifestyle,
diabetes, and cardiovascular risk factors 10 years after bariatric
surgery. N Engl J Med. 351:2683–2693. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Barres R and Zierath JR: DNA methylation
in metabolic disorders. Am J Clin Nutr. 93:897S–900S. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Takizawa T, Nakashima K, Namihira M, et
al: DNA methylation is a critical cell-intrinsic determinant of
astrocyte differentiation in the fetal brain. Dev Cell. 1:749–758.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Robertson KD: DNA methylation and human
disease. Nat Rev Genet. 6:597–610. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rönn T, Poulsen P, Hansson O, et al: Age
influences DNA methylation and gene expression of COX7A1 in human
skeletal muscle. Diabetologia. 51:1159–1168. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ling C, Poulsen P, Carlsson E, et al:
Multiple environmental and genetic factors influence skeletal
muscle PGC-1α and PGC-1β gene expression in twins. J Clin Invest.
114:1518–1526. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ling C, Poulsen P, Simonsson S, et al:
Genetic and epigenetic factors are associated with expression of
respiratory chain component NDUFB6 in human skeletal muscle. J Clin
Invest. 117:3427–3435. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jensen CB, Storgaard H, Madsbad S, Richter
EA and Vaag AA: Altered skeletal muscle fiber composition and size
precede whole-body insulin resistance in young men with low birth
weight. J Clin Endocrinol Metab. 92:1530–1534. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Barrett T, Wilhite SE, Ledoux P, et al:
NCBI GEO: archive for functional genomics data sets - update.
Nucleic Acids Res. 41:D991–D995. 2013. View Article : Google Scholar
|
|
17
|
Du P, Zhang X, Huang CC, et al: Comparison
of Beta-value and M-value methods for quantifying methylation
levels by microarray analysis. BMC Bioinformatics. 11:5872010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Da Wei Huang BTS, Sherman BT and Lempicki
RA: Systematic and integrative analysis of large gene lists using
DAVID bioinformatics resources. Nat Protoc. 4:44–57. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dubchak I, Munoz M, Poliakov A, et al:
Whole-Genome rVISTA: a tool to determine enrichment of
transcription factor binding sites in gene promoters from
transcriptomic data. Bioinformatics. 29:2059–2061. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wingender E, Dietze P, Karas H and Knüppel
R: TRANSFAC: A database on transcription factors and their DNA
binding sites. Nucleic Acids Res. 24:238–241. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kume S, Uzu T, Kashiwagi A and Koya D:
SIRT1, a calorie restriction mimetic, in a new therapeutic approach
for type 2 diabetes mellitus and diabetic vascular complications.
Endocr Metab Immune Disord Drug Targets. 10:16–24. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sharma S, Misra CS, Arumugam S, et al:
Antidiabetic activity of resveratrol, a known SIRT1 activator in a
genetic model for type 2 diabetes. Phytother Res. 25:67–73. 2011.
View Article : Google Scholar
|
|
23
|
Bordone L, Motta MC, Picard F, et al:
Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic
β cells. PLoS Biol. 4:e312006. View Article : Google Scholar
|
|
24
|
Bhakta S, Besra GS, Upton AM, et al:
Arylamine N-acetyltransferase is required for synthesis of mycolic
acids and complex lipids in Mycobacterium bovis BCG and represents
a novel drug target. J Exp Med. 199:1191–1199. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Aljakna A, Choi S, Savage H, et al:
Pla2g12b and Hpn are genes identified by mouse ENU mutagenesis that
affect HDL cholesterol. PLoS One. 7:e431392012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Blum M, Grant DM, Mcbride W, Heim M and
Meyer UA: Human arylamine N-acetyltransferase genes: isolation,
chromosomal localization, and functional expression. DNA Cell Biol.
9:193–203. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Heit JJ, Apelqvist AA, Gu X, et al:
Calcineurin/NFAT signalling regulates pancreatic β-cell growth and
function. Nature. 443:345–349. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Papadopoulou V, Postigo A, Sánchez-Tilló
E, Porter AC and Wagner SD: ZEB1 and CtBP form a repressive complex
at a distal promoter element of the BCL6 locus. Biochem J.
427:541–550. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Karnik SK, Chen H, McLean GW, et al: Menin
controls growth of pancreatic β-cells in pregnant mice and promotes
gestational diabetes mellitus. Science. 318:806–809. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nan X, Ng HH, Johnson CA, et al:
Transcriptional repression by the methyl-CpG-binding protein MeCP2
involves a histone deacetylase complex. Nature. 393:386–389. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li E, Beard C and Jaenisch R: Role for DNA
methylation in genomic imprinting. Nature. 366:362–365. 1993.
View Article : Google Scholar : PubMed/NCBI
|