Introduction
Oral squamous cell carcinoma (OSCC) is responsible
for 24% of all cases of head and neck cancer (1). Despite advances in multimodality
treatment, the overall prognosis for patients with OSCC,
particularly tongue cancer, has changed little in the last three
decades (2). Furthermore, the
reasons for variability in the clinical progression of patients
with tongue squamous cell cancer (TSCC) remain to be elucidated.
Identification of novel prognostic factors may enable the rational
selection of appropriate therapeutic options for individual
patients. Transcriptional profiling by DNA microarray analysis is
an effective tool in cancer research, significantly improving
current knowledge regarding tumor development and progression
(3). It has also assisted in
identifying novel treatment targets and to generate prediction
models for prognosis and treatment response (4–6). The
cellular and molecular heterogeneity of OSCC, and the numerous
genes potentially involved in its development, reiterate the
importance of investigating gene alterations on a global scale.
MicroRNAs (miRNAs) are a class of non-coding RNAs,
which are between 21 and 25 nucleotides in length and function as
regulators of gene expression. Mature miRNAs and Argonaute proteins
form the RNA-induced silencing complex, which mediates
post-transcriptional gene silencing through induction of mRNA
degradation or the inhibition of translation (7). It has previously been estimated that
one third of the genes in the human genome are regulated by miRNAs
(8), and >1,800 miRNAs have
been identified in miRBase version 20.0 (9). miRNAs are involved in several key
cellular processes, including apoptosis, proliferation and
differentiation (10).
Dysregulaton in the expression of miRNA or miRNA mutations result
in a gain or loss of miRNA function, which leads to downregulation
or upregulation of the target protein. miRNAs have also been found
to function as oncogenes or tumor suppressors (11,12).
However, there have been few reports regarding the
role of miRNAs in the regulation of TSCC. Furthermore, the
regulation of miRNAs and corresponding target mRNAs during the
occurrence and development of TSCC remains to be elucidated. The
introduction of genome-wide technologies, including gene expression
microarrays, has made it possible to achieve a comprehensive view
of the alterations of miRNAs and mRNAs involved in TSCC. In
addition, the use of bioinformatics enables the analysis of the
differentially expressed miRNAs and mRNAs.
The present study aimed to identify the miRNAs and
mRNAs involved in the molecular changes associated with TSCC.
Published gene expression microarray databases of miRNAs and mRNAs
were examined, in order to discriminate between the expression
profiles of TSCC samples and normal control samples. The aim was to
determine whether the regulation of miRNAs is involved in the
pathophysiology of TSCC, and identify novel mechanisms and targets
for cancer therapy.
Patients and methods
Patients
Microarray data were obtained from three datasets,
which consisted of 18, 57 and 38 appropriate samples, respectively.
The miRNA microarray series contained data from 15 tumor samples
and three healthy control samples, the mRNA microarray test series
contained data from 26 tumor samples and 12 healthy control
samples, and the mRNA microarray confirmation series contained data
from 37 tumor samples and 20 healthy control samples. The three
series were accessed at the National Centers for Biotechnology
Information (NCBI) Gene Expression Omnibus (GEO) database
(http://www.ncbi.nlm.nih.gov/geo/), and
the accession numbers were GSE28100, GSE9844 and GSE13601,
respectively. Details of the sample features have been presented in
previous reports (13–15). The raw values of miRNAs were
collected from microarray data and normalized by logarithmic
transformation for the ease of further calculation.
Differentially expressed miRNAs
Differentially expressed miRNAs between the TSCC and
normal control samples were identified using the limma method. The
P-value and false discovery rate (FDR) were calculated for each
differentially expressed miRNA. A threshold was set at
fold-change>4, P<0.01 and FDR<0.01, from which the
TSCC-associated differentially expressed miRNAs were selected.
Unsupervised hierarchical clustering was performed with Cluster
software, version 3.0 (Eisen Lab, Stanford, CA, USA) using
Pearson’s correlation distance metric and average linkage. The
cluster was visualized using Treeview software (Eisen Lab)
(16).
Differentially expressed mRNAs
The differentially expressed mRNAs between the TSCC
and normal control samples were identified using the limma method
(17). The P-value and the
fold-change were calculated for each differentially expressed mRNA.
Thresholds were set at P<0.01 and FDR<0.01, from which the
TSCC-associated differentially expressed mRNAs were selected.
Unsupervised hierarchical clustering was performed with Cluster
software using Pearson’s correlation distance metric and average
linkage, followed by visualization using Treeview software.
Gene Ontology (GO) analysis
GO analysis was used to analyze the predominant
functions of the differentially expressed genes according to the
GO, which is the key functional classification of the NCBI
(18,19). Fisher’s exact test and
χ2 test were used to classify the GO category,
and the FDR (20) was calculated
to correct the P-value; the smaller the FDR, the smaller the error
in judging the P-value. The FDR was defined as FDR = 1 -
Nk / T, where Nk refers to the
number of Fisher’s test P-values less than the χ2
test P-values. P-values were calculated for the GO terms of all the
differentially expressed genes. Enrichment provides a measure of
the significance of the function; as the enrichment increases, the
corresponding function is considered more specific, enabling
identification of the GO terms with more significant functions
(21).
Pathway analysis
Pathway Analysis (The Intelligent Systems and
Bioinformatics Laboratory, Detroit, MI, USA) was used to identify
the significant pathways associated with the differentially
expressed genes, according to Kyoto Encyclopedia of Genes and
Genomes (KEGG; http://www.genome.jp/kegg/), Biocarta (http://www.biocarta.com/) and Reatome (http://www.reactome.org/). Fisher’s exact test and
χ2 test were used to select the significant
pathways, and the threshold of significance was defined by the
P-value (<0.05) and FDR (<0.05) (22–24).
Annotation of the miRNA targets
The target mRNAs of the differentially expressed
miRNAs were predicted based on TargetScan (http://www.targetscan.org/) version 5.2. TargetScan
predicts the biological targets of miRNAs by identifying conserved
8mer and 7mer sites, which match the seed region of each miRNA
(8). Sites containing mismatches
in the seed region, which are compensated by conserved 3′ pairing,
are also identified (25). In
mammals, predictions are ranked based on the predicted efficacy of
targeting, as calculated using the context scores of the site
alignments (26,27). TargetScan Human considers matches
to annotate human untranslated regions and their orthologs, as
defined by UCSC whole-genome alignments. Conserved targeting is
also detected within open reading frames.
miRNA-GO network
The miRNA-GO network was generated according to the
association between significant GO terms and miRNAs. The adjacency
matrix of miRNA and GO terms: A=(ai,j) was calculated
from the association between the GO terms and the microRNAs, where
ai,j represents the association weight of GO (i) and
microRNA (j). In the miRNA-GO network, squares represent microRNAs
and circles represent GO terms, and their association is
represented by one edge. The center of the network was defined by
the degree, which was the contribution that one microRNA made to
the surrounding GO terms, or the contribution that one GO term made
to the surrounding microRNAs. The key microRNA and GO term in the
network always have the highest degrees (28).
miRNA-mRNA network
The association between differentially expressed
miRNAs and mRNAs were calculated by their differential expression
values. The network was generated, according to the interactions
between the miRNAs and mRNAs listed in the Sanger microRNA database
(http://www.mirbase.org/). The adjacency matrix of
the miRNA and mRNAs was calculated using A=(ai,j), as
described above, where ai,j represents the association
weight of the mRNA (i) and miRNA (j). The degree was defined as the
contribution one miRNA made to the surrounding mRNAs, or the
contribution one gene made to the surrounding miRNAs. The key miRNA
and gene in the network always have the highest degrees.
Statistical analysis
The numerical data are presented as the mean ±
standard deviation. Differences between the means were analyzed
using Student’s t-test. All statistical analyses were performed
using SPSS version 13.0 software (SPSS, Inc,. Chicago, IL,
USA).
Results
Overview of the miRNA expression
profiles
From the miRNA expression profiles, differentially
expressed miRNAs were identified between the TSCC and normal
control samples. The miRNA expression profiles in the TSCC samples
were determined by calculating the log fold-change TSCC/normal.
Since the sample size was limited, the fold-change, FDR and
P-values were calculated from the normalized expression values and
26 results were obtained. According to the miRBase (http://www.mirbase.org/) database, miRNA-923 was
observed to not be a true miRNA, thus was excluded from further
investigation. Therefore, 25 differentially expressed human miRNAs
were identified between the 15 TSCC patients and three normal
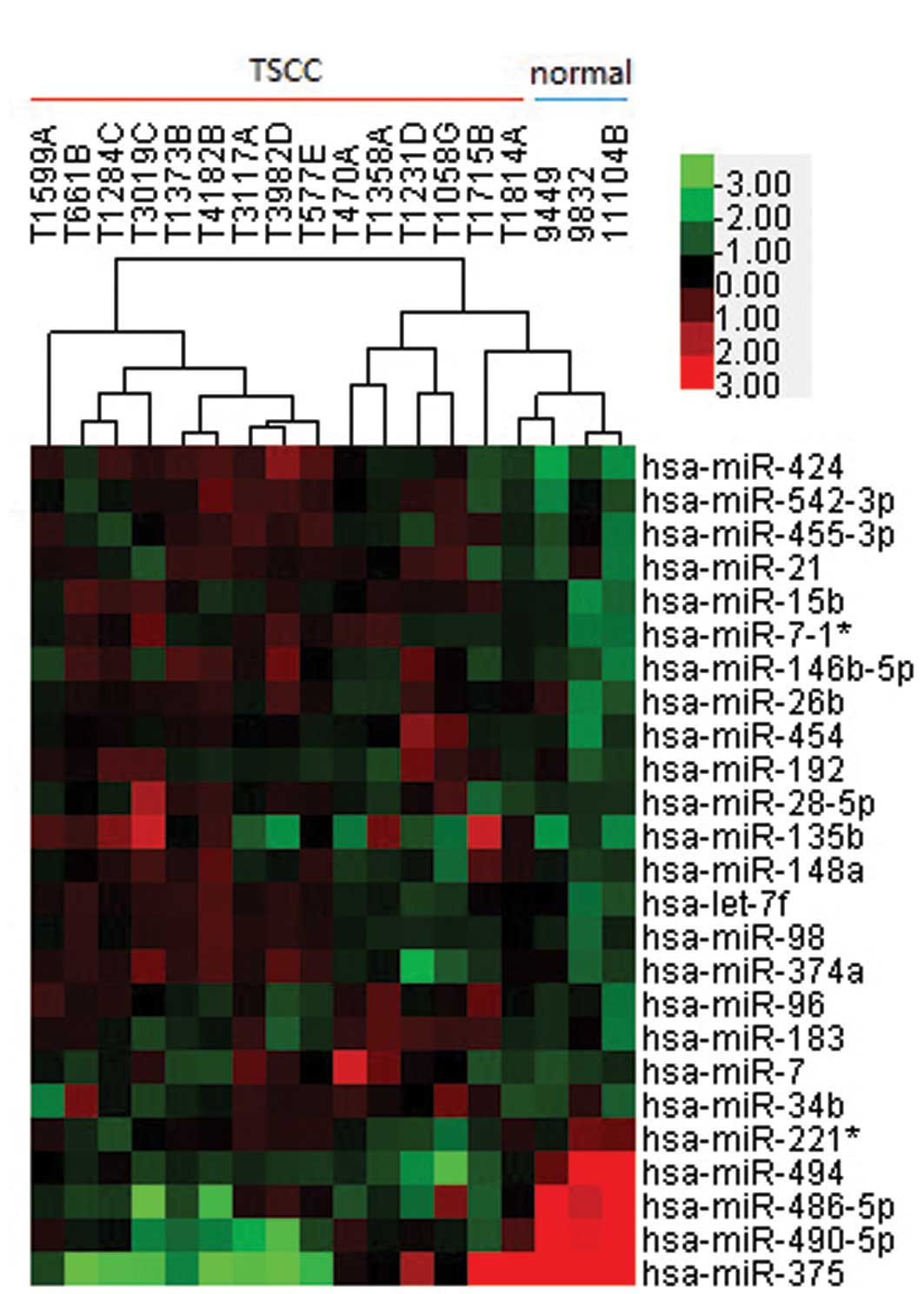
controls. A heat map, constructed using unsupervised hierarchical
clustering analyses with threshold values set at fold-change >2,
P<0.01 and FDR<0.01, demonstrated that there were 21
overexpressed and four underexpressed miRNAs in the TSCC samples,
compared with the normal tissue samples (Fig. 1). According to the FDR values,
miR-424, miR-542-3p and miR-454 were the most upregulated and
miR-494, miR-490-5p and miR-486-5p were the most downregulated
miRNAs, compared with the normal tissue samples (Table I). Upregulated miRNAs were more
common, compared with downregulated miRNAs in the TSCC group.
 | Table IDifferentially expressed miRNAs
detected by microarray analysis of TSCC samples. |
Table I
Differentially expressed miRNAs
detected by microarray analysis of TSCC samples.
| miRNA | P-value | Fold-change |
|---|
| Downgulated in
TSCC | | |
|
hsa-miR-490-5p | 4.84E-04 | 0.22 |
| hsa-miR-494 | 1.54E-04 | 0.17 |
|
hsa-miR-486-5p | 2.01E-03 | 0.21 |
| hsa-miR-375 | 3.17E-03 | 0.07 |
| Upregulated in
TSCC | | |
| hsa-miR-424 | 2.36E-06 | 9.55 |
| hsa-miR-454 | 7.29E-06 | 5.50 |
|
hsa-miR-542-3p | 1.10E-05 | 5.67 |
| hsa-miR-15b | 1.40E-05 | 6.58 |
Overview of the mRNA expression
profiles
In the mRNA microarray test group, ≤54,675 coding
transcripts were detected in the 38 samples, using the Affymetrix
U133 plus 2.0 array (Affymetrix, Santa Clara, CA, USA). Using the
limma method, with a cut-off value of FDR<0.01 between the two
groups, 324 probes were upregulated in the TSCC samples and 445
were downregulated. Global mRNA expression patterns were evaluated
using hierarchical clustering. The most differentially expressed
mRNAs revealed two major clusters, which correlated with the
differentiation state of the tumor (Fig. 2). Expression cluster 1 contained
all 12 normal control samples and three TSCC samples, whereas
expression cluster 2 contained 23 of the 26 TSCC samples. Matrix
metalloproteinase 1 was the most significantly upregulated mRNA,
and tenascin XB was the most significantly downregulated mRNA
(Table II). Downregulated mRNAs
were more common than upregulated mRNAs in the TSCC group.
| Table IIDifferentially expressed mRNAs
detected by microarray analysis of TSCC samples. |
Table II
Differentially expressed mRNAs
detected by microarray analysis of TSCC samples.
| mRNA | P-value | Fold-change |
|---|
| Downregulated in
TSCC | | |
| TNXB | 1.14E-11 | 0.20 |
| PADI1 | 6.44E-11 | 0.18 |
| SPNS2 | 2.22E-10 | 0.27 |
| CD1C | 6.93E-10 | 0.27 |
| GSTM5 | 1.99E-09 | 0.34 |
| ADH1B | 2.55E-09 | 0.07 |
| HLF | 3.12E-09 | 0.37 |
| HPGD | 4.38E-09 | 0.11 |
| GCOM1 | 6.39E-09 | 0.17 |
| BBIP1 | 7.96E-09 | 0.48 |
| Upregulated in
TSCC | | |
| MMP1 | 4.44E-16 | 123.60 |
| MYO1B | 3.10E-11 | 4.40 |
| IL8 | 1.64E-10 | 10.08 |
| PTHLH | 5.93E-10 | 17.09 |
| MYO1B | 9.47E-10 | 5.15 |
| MMP3 | 1.02E-09 | 10.48 |
| CDH3 | 6.25E-09 | 5.33 |
| KRT17 | 8.74E-09 | 13.53 |
| COL4A6 | 1.08E-08 | 6.89 |
| CXCL1 | 1.44E-08 | 10.85 |
Microarray-based GO analysis
The target mRNAs of the differentially expressed
miRNAs were predicted using TargetScan (http://www.targetscan.org/). A total of 5,208
associations between the mRNAs and miRNAs were observed. The
intersection set for the predicted target mRNAs and the
differentially expressed mRNAs identified from the GSE13601 dataset
were selected. Following negative correlation analysis, the
eligible mRNAs underwent GO analysis. P<0.01 was considered to
indicate GO terms, which were significantly regulated by the
differentially expressed miRNAs. The characteristics and
associations between the miRNAs and mRNAs are listed in Table III. The highly enriched GO terms
targeted by the miRNAs, included regulation of transcription,
development and cell differentiation The maximum-enriched GO term
was signal transduction, which is a known to be a basic function of
miRNA. An miRNA-GO network was constructed to indicate the GO
terms, which function in the regulation of TSCC (Fig. 3). In the network, miRNA-494,
miRNA-183 and miRNA-96 were found to be central in regulating the
majority of the GO terms.
| Table IIImiRNA-gene ontology network
characteristics. |
Table III
miRNA-gene ontology network
characteristics.
miRNA
| Gene Ontology
|
|---|
| Name | Degree | Term | Degree |
|---|
| hsa-miR-494 | 22 | Regulation of
transcription, DNA-dependent | 12 |
| hsa-miR-183 | 14 | Transcription | 9 |
| hsa-miR-96 | 11 | Development | 8 |
| hsa-miR-374a | 10 | Signal
transduction | 6 |
| hsa-miR-21 | 6 | Apoptosis | 5 |
| hsa-miR-455-3p | 6 | Cell
differentiation | 5 |
| hsa-miR-542-3p | 6 | Cell cycle | 4 |
|
hsa-miR-490-5p | 5 | Cell cycle
arrest | 4 |
| hsa-miR-15b | 4 | Cell division | 4 |
| hsa-miR-7 | 4 | Negative regulation
of progression through cell cycle | 3 |
| hsa-let-7f | 3 | Positive regulation
of transcription from RNA polymerase II promoter | 3 |
| hsa-miR-98 | 3 | Rhythmic
process | 3 |
| hsa-miR-192 | 2 | Cell aging | 2 |
| hsa-miR-34b | 2 | Cytokinesis | 2 |
| hsa-miR-375 | 2 | Negative regulation
of JNK activity | 2 |
| | Negative regulation
of transcription from RNA polymerase II promoter | 2 |
| | Organic anion
transport | 2 |
| | Protein amino acid
phosphorylation | 2 |
| | Response to
hypoxia | 2 |
| | Transcription from
RNA polymerase II promoter | 2 |
| | Actin
modification | 1 |
| | Cell
dedifferentiation | 1 |
| | Cellular response
to insulin stimulus | 1 |
| | Chromatin
remodeling | 1 |
| | G1 phase of mitotic
cell cycle | 1 |
| | Glucose
homeostasis | 1 |
| | Insulin-like growth
factor receptor signaling pathway | 1 |
| | Leukemia inhibitory
factor signaling pathway | 1 |
| | NAD metabolism | 1 |
| | Negative regulation
of epithelial cell proliferation | 1 |
| | Negative regulation
of insulin-like growth factor receptor signaling pathway | 1 |
| | Phosphoinositide
3-kinase cascade | 1 |
| | Positive regulation
of fibroblast proliferation | 1 |
| | Positive regulation
of glucose import | 1 |
| | Positive regulation
of mitotic metaphase/anaphase transition | 1 |
| | Protein kinase B
signaling cascade | 1 |
| | Response to
cytokine stimulus | 1 |
| | Response to peptide
hormone stimulus | 1 |
Microarray-based pathway analysis
The results of the present study suggested that
signal transduction and other GO terms may be involved in TSCC,
therefore, associated pathways were analyzed, according to the
functions and interactions of the differentially expressed genes.
Pathway analysis considers relative change direction and
fold-change, and the threshold of significance is P<0.05
(29). Using pathway analysis, 14
significant pathways were identified (Fig. 4). The highly enriched pathways
targeted by dysregulated mRNAs were: Peroxisome
proliferator-activated receptor (PPAR) signaling pathway, adherens
junction and extracellular cell membrane (ECM)-receptor
interaction. These results suggested that miRNAs may regulate the
oncogenesis of TSCC through these pathways.
MicroRNA-mRNA network
The overlapping mRNAs from the TargetScan
predictions and the results of the mRNA microarray of
differentially expressed mRNAs in the GO and pathway analyses were
selected. The miRNA-mRNA regulatory networks based on these mRNAs
were used to identify the putative target mRNAs of the
overexpressed and underexpressed miRNAs (Fig. 5). The total number of mRNAs and
miRNAs in the network were 68 and 19, respectively. The
associations between the miRNAs and the mRNAs are listed in
Table IV. In the network,
circular nodes indicated mRNAs, square nodes were miRNAs, and edges
between two nodes indicated the interactions between the miRNAs and
mRNAs. The degree represents the number of target genes regulated
by a certain miRNA. The higher the degree, the more central the
miRNA occurs within the network. Three dysregulated miRNAs
(miR-494, miR-96, and miR-455-3p) had the highest number of target
mRNAs. In addition, runt-related transcription factor 1 (RUNX1T1),
alkylglycerone phosphate synthase and cyclin-dependent kinase
(CDK)19 were targeted by the highest number of miRNAs.
| Table IVmiR-mRNA network characteristics. |
Table IV
miR-mRNA network characteristics.
| Target | Degree |
|---|
| miRNA |
| hsa-miR-494 | 14 |
| hsa-miR-96 | 11 |
|
hsa-miR-455-3p | 8 |
|
hsa-miR-542-3p | 7 |
| hsa-miR-183 | 6 |
| hsa-miR-374a | 6 |
| hsa-miR-375 | 5 |
| hsa-miR-21 | 4 |
| hsa-miR-7 | 4 |
| hsa-miR-148a | 3 |
| hsa-miR-192 | 3 |
| hsa-miR-221 | 3 |
| hsa-miR-34b | 3 |
|
hsa-miR-490-5p | 3 |
| hsa-let-7f | 2 |
| hsa-miR-15b | 2 |
| hsa-miR-424 | 2 |
| hsa-miR-454 | 1 |
| hsa-miR-98 | 1 |
| mRNA |
| RUNX1T1 | 3 |
| AGPS | 2 |
| CDK19 | 2 |
| ENAH | 2 |
| IKZF2 | 2 |
| MAGI1 | 2 |
| MEIS1 | 2 |
| NEBL | 2 |
| NFIA | 2 |
| PAQR8 | 2 |
| PDCD4 | 2 |
| PLCXD3 | 2 |
| PLEKHA6 | 2 |
| PTGER3 | 2 |
| RECK | 2 |
| SESN1 | 2 |
| SH3BGRL2 | 2 |
| SLC16A7 | 2 |
| TACC1 | 2 |
| ATOH8 | 1 |
| C18orf25 | 1 |
| CAPN5 | 1 |
| CBX3 | 1 |
| CCNG2 | 1 |
| CCRN4L | 1 |
| CDK6 | 1 |
| CHRDL1 | 1 |
| CILP | 1 |
| CKS1B | 1 |
| CLPTM1L | 1 |
| DIAPH2 | 1 |
| ESPL1 | 1 |
| FBXO45 | 1 |
| FCHO2 | 1 |
| GALNT2 | 1 |
| GAS7 | 1 |
| HLF | 1 |
| IRS1 | 1 |
| ITGA9 | 1 |
| ITPR2 | 1 |
| KIAA0141 | 1 |
| KLB | 1 |
| LIFR | 1 |
| LYRM7 | 1 |
| MBNL3 | 1 |
| MRPS23 | 1 |
| MYRIP | 1 |
| NADK | 1 |
| NCF2 | 1 |
| NCOA1 | 1 |
| NR2F2 | 1 |
| NTRK3 | 1 |
| PER3 | 1 |
| PHF17 | 1 |
| PLAGL1 | 1 |
| PPFIA1 | 1 |
| RUFY3 | 1 |
| SAMD5 | 1 |
| SLC16A6 | 1 |
| SLC7A11 | 1 |
| SLIT3 | 1 |
| ST3GAL3 | 1 |
| TACR1 | 1 |
| TOR1A | 1 |
| TRIOBP | 1 |
| VSIG10 | 1 |
| WDR26 | 1 |
| ZNF281 | 1 |
miRNAs may exhibit their central involvement through
the above mRNAs, and regulate the formation and development of
TSCC. In the present study, another mRNA expression profile data
series was used for confirmation of these findings.
GSE13601 was used as the confirmation data series,
which contained 58 samples. The ‘core’ mRNAs in the miRNA-mRNA
network were assessed based on the same screening criteria and an
associated heat map was constructed. Among the 68 originally
identified mRNAs, 45 mRNAs were detected in the GSE13601 data serie
using an Affymetrix U95 version 2 array. The TSCC samples were
successfully discriminated from the normal control samples in this
series (Fig. 6). Furthermore, the
fold-changes of these mRNAs in GSE13601 were calculated, and 42 of
the 45 mRNAs exhibited the same change direction as the GSE9844
data series. Only SLC7A11, CDK6 and CDK19 exhibited
differences.
Discussion
Despite advances in surgery and radiation therapy,
the 5-year survival rate for oral cancer has not improved
significantly for several decades and remains at 50–55% (30,31).
At present, numerous novel prognostic factors, including
cytological features, standard karyotyping, fluorescence in
situ hybridization, centromeric probes, single nucleotide
polymorphism and gene expression profiling are being investigated.
Following technical advances and reductions in the cost of gene
expression microarrays, they are considered a useful tool for
investigating the development and progression of tumors. Owing to
the high throughout of microarrays, novel genes that affect the
development of TSCC can be identified. miRNAs are regulatory
factors, which are considered to be involved in the progression of
TSCC and provide a possible target for TSCC therapy (13).
Understanding the clinical relevance of miRNA
expression patterns in TSCC is necessary to classify heterogeneous
tumors and circumvent the therapeutic challenges faced in their
clinical management. However, miRNAs, which indirectly regulate the
pathophysiological process of TSCC, and the possible target mRNAs,
require elucidation. Microarrays are a useful tool for
investigating the development and progression of tumors, owing to
their high throughout; however, it remains difficult to predict
TSCC, predominantly due to the challenges in interpreting the
complex data produced (32) and
determining the responsible genes. The present study used
bioinformatics to analyze the functions and pathways associated
with differentially expressed miRNAs and mRNAs, to further clarify
their biological significance to reveal the key miRNAs and possible
target mRNAs affecting the formation of TSCC.
The present study identified 26 differentially
expressed miRNAs in TSCC compared with normal tongue tissue
samples. Since the expression of miRNA is known to be tissue- and
tumor-specific (33), using the
appropriate subset of tumor samples and the corresponding normal
control samples is important to reduce the potential complexities
associated with analyzing heterogeneous tumors. The present study
aimed to investigate miRNA-mRNA regulation in TSCC, therefore, two
gene expression microarray datasets were used to identify the mRNA
targets of miRNAs. The GSE9844 dataset was used as the test
expression profile, in which 769 differently expressed mRNAs were
identified. The mRNAs, which were negatively correlated with the
previously identified differentially expressed miRNAs were then
used to further investigate the role of miRNAs in TSCC.
The GO is widely recognized as the leading tool for
the organization and functional annotation of molecular attributes
(34). By using a cut-off value of
P<0.01, significant GO terms and associated genes were
identified. Guo et al (35)
previously performed a GO analysis to analyze an miRNA microarray,
and revealed that mir-15b and miR-16 may be indispensable for
apoptosis through targeting B-cell lymphoma 2. In the present
study, GO terms for the transcriptional regulatory response were
found to be important in TSCC through the function of miRNAs. This
finding was concordant with the predominant biological function of
miRNAs in humans. Transcriptional regulation is a major function of
miRNAs (36), and the significant
changes in this term in the present study further certified the
results. Jiang et al (37)
previously reported that miRNA-7 contributes to the suppression of
tumorigenesis in TSCC by targeting insulin-like growth factor 1
through cell cycle arrest (37).
In addition, Yao et al (38) demonstrated that sulforaphane
inhibits hypoxia-inducible factor-1α by activating the c-Jun
N-terminal kinase pathway in TSCC (38). Therefore, it was hypothesized that
the other miRNAs listed may have functions in the progression of
TSCC, which remain to be elucidated.
Pathway analyses can reveal the distinct biological
processes and significant pathways associated with the
differentially expressed mRNAs. This permits a comprehensive
understanding of the interactions of genes, the functions that they
are involved in, and associations between upstream and downstream
genes. Pathway analyses can also identify genes associated with
these significant pathways, which may be regulated by miRNAs. The
present study identified pathways regarding PPAR signaling,
adherens junctions and p53 signaling, thus confirming their
concordance with GO terms and their importance in TSCC. The PPAR
signaling pathway has previously been considered a useful
prognostic marker and a potential therapeutic target for TSCC
(39), however, there have been no
reports regarding its molecular mechanism and miRNA regulation.
Numerous studies have demonstrated that cytokine interaction is
involved in the process of tumor growth, which is important for
TSCC (40,41). The miR-34a-sirtuin 6 axis was
previously found to be involved in various types of squamous cell
cancer, which also demonstrates the impact of the p53 signaling
pathway on TSCC (42). There
remains a lack of information regarding miRNAs in TSCC or the
associated signaling pathway information regulated by miRNAs. The
results of the present study suggested that other, apparently
irrelevant pathways were be controlled by miRNAs and have functions
in TSCC, which requires further investigation. In the present
study, the pathway analyses identified equally important roles and
functions as the GO analysis.
The present study also investigated the genes
associated with significant GO terms and pathways, and 68 genes in
common were identified, possibly regulated by miRNAs in TSCC. It
has been demonstrated that miRNA-494 is upregulated in whole blood
samples of patients with OSCC and may be used as a potential
biomarker (43) however, there
remains a lack of information regarding its role in TSCC. miRNA-96
and -183 belong to the miRNA-183 family and, in a meta-analysis
have been identified as useful prognostic markers and therapeutic
targets in various types of cancer (44). The RUNX family includes
sequence-specific transcription factors, which are closely
associated with various cellular processes, including development,
differentiation and/or tumorigenesis, and have been implicated in
cancer cells through the miRNA-23a cluster (45). PDCD4 has been also been suggested
as a potential marker of TSCC, as selected by a cDNA microarray
(46). Membrane-associated
guanylate kinase (MAGI1) is a partner of the PDZ-domain and has
been also been identified as a serum biomarker of OSCC (47). Although their functions remain to
be fully elucidated, several miRNAs may be associated with the
regulation of TSCC. In the present study, the second mRNA
expression series, used for confirmation, verified the accuracy of
the initial results. Based on these data, further investigation of
miRNA expression and target functions, and investigation of the
regulation of the identified miRNAs and pathway functions are
required. This may assist in improving the clinical diagnosis and
treatment of patients with TSCC.
In conclusion, the present study identified, by
correlating the mRNA and miRNA expression data from two different
platforms, putative miRNA-mRNA interactions in TSCC. The miRNA-GO
network and the miRNA-pathway analyses identified pathways
controlling the PPAR and p53 signaling pathways and adherens
junctions, and the focal adhesion and ECM-receptor interaction
pathways. Network analysis identified important miRNAs and mRNAs,
including miRNA-494, miRNA-96, miRNA-183, RUNX1T1, PDCD4 and MAGI1,
which may be involved in the progression of TSCC. The successful
verification of these mRNAs in the GSE13601 series provided further
evidence that differentially expressed miRNAs in TSCC may regulate
tumor formation through regulation of target mRNAs, and may be used
to discriminate tumor samples from normal samples. Based on the
integrated analysis of transcription features, these results may
provide an important contribution to future investigations aimed at
characterizing the role of specific miRNAs in the pathogenesis of
TSCC, and may contribute to improving diagnosis and treatment.
References
|
1
|
Parkin DM, Pisani P and Ferlay J: Global
cancer statistics. CA Cancer J Clin. 49:33–64. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Arya S, Rane P and Deshmukh A: Oral cavity
squamous cell carcinoma: role of pretreatment imaging and its
influence on management. Clin Radiol. 69:916–930. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
D’Angelo G, Di Rienzo T and Ojetti V:
Microarray analysis in gastric cancer: a review. World J
Gastroenterol. 20:11972–11976. 2014. View Article : Google Scholar
|
|
4
|
Chon HS and Lancaster JM: Microarray-based
gene expression studies in ovarian cancer. Cancer Control. 18:8–15.
2011.PubMed/NCBI
|
|
5
|
Vitucci M, Hayes DN and Miller CR: Gene
expression profiling of gliomas: merging genomic and
histopathological classification for personalised therapy. Br J
Cancer. 104:545–553. 2011. View Article : Google Scholar :
|
|
6
|
Nannini M, Pantaleo MA, Maleddu A, Astolfi
A, Formica S and Biasco G: Gene expression profiling in colorectal
cancer using microarray technologies: results and perspectives.
Cancer Treat Rev. 35:201–209. 2009. View Article : Google Scholar
|
|
7
|
Lin PY, Yu SL and Yang PC: MicroRNA in
lung cancer. Br J Cancer. 103:1144–1148. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lewis BP, Burge CB and Bartel DP:
Conserved seed pairing, often flanked by adenosines, indicates that
thousands of human genes are microRNA targets. Cell. 120:15–20.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kozomara A and Griffiths-Jones S: miRBase:
integrating microRNA annotation and deep-sequencing data. Nucleic
Acids Res. 39:D152–157. 2011. View Article : Google Scholar :
|
|
10
|
Li X, Zhang J, Gao L, et al: MiR-181
mediates cell differentiation by interrupting the Lin28 and let-7
feedback circuit. Cell Death Differ. 19:378–386. 2012. View Article : Google Scholar :
|
|
11
|
Lee YS and Dutta A: MicroRNAs: small but
potent oncogenes or tumor suppressors. Curr Opin Investig Drugs.
7:560–564. 2006.PubMed/NCBI
|
|
12
|
Caldas C and Brenton JD: Sizing up miRNAs
as cancer genes. Nat Med. 11:712–714. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Estilo CL, O-charoenrat P, Talbot S, et
al: Oral tongue cancer gene expression profiling: Identification of
novel potential prognosticators by oligonucleotide microarray
analysis. BMC Cancer. 9:112009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ye H, Yu T, Temam S, et al: Transcriptomic
dissection of tongue squamous cell carcinoma. BMC Genomics.
9:692008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jung HM, Phillips BL, Patel RS, et al:
Keratinization-associated miR-7 and miR-21 regulate tumor
suppressor reversion-inducing cysteine-rich protein with kazal
motifs (RECK) in oral cancer. J Biol Chem. 287:29261–29272. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Eisen MB, Spellman PT, Brown PO and
Botstein D: Cluster analysis and display of genome-wide expression
patterns. Proc Natl Acad Sci USA. 95:14863–14868. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Diboun I, Wernisch L, Orengo CA and
Koltzenburg M: Microarray analysis after RNA amplification can
detect pronounced differences in gene expression using limma. BMC
Genomics. 7:2522006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gene Ontology Consortium: The Gene
Ontology (GO) project in 2006. Nucleic Acids Res. 34:D322–D326.
2006. View Article : Google Scholar
|
|
19
|
Ashburner M, Ball CA, Blake JA, et al:
Gene ontology: tool for the unification of biology. The Gene
Ontology Consortium. Nat Genet. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dupuy D, Bertin N, Hidalgo CA, et al:
Genome-scale analysis of in vivo spatiotemporal promoter activity
in Caenorhabditis elegans. Nat Biotechnol. 25:663–668. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schlitt T, Palin K, Rung J, et al: From
gene networks to gene function. Genome Res. 13:2568–2576. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kanehisa M, Goto S, Kawashima S, Okuno Y
and Hattori M: The KEGG resource for deciphering the genome.
Nucleic Acids Res. 32:D277–D280. 2004. View Article : Google Scholar :
|
|
23
|
Yi M, Horton JD, Cohen JC, Hobbs HH and
Stephens RM: WholePathwayScope: a comprehensive pathway-based
analysis tool for high-throughput data. BMC Bioinformatics.
7:302006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Draghici S, Khatri P, Tarca AL, et al: A
systems biology approach for pathway level analysis. Genome Res.
17:1537–1545. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Friedman RC, Farh KK, Burge CB and Bartel
DP: Most mammalian mRNAs are conserved targets of microRNAs. Genome
Res. 19:92–105. 2009. View Article : Google Scholar :
|
|
26
|
Grimson A, Farh KK, Johnston WK,
Garrett-Engele P, Lim LP and Bartel DP: MicroRNA targeting
specificity in mammals: determinants beyond seed pairing. Mol Cell.
27:91–105. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Garcia DM, Baek D, Shin C, Bell GW,
Grimson A and Bartel DP: Weak seed-pairing stability and high
target-site abundance decrease the proficiency of lsy-6 and other
microRNAs. Nat Struct Mol Biol. 18:1139–1146. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang W, Edwards A, Fan W, Flemington EK
and Zhang K: miRNA-mRNA correlation-network modules in human
prostate cancer and the differences between primary and metastatic
tumor subtypes. PLoS One. 7:e401302012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang Y, Li H, Hou S, Hu B, Liu J and Wang
J: The noncoding RNA expression profile and the effect of lncRNA
AK126698 on cisplatin resistance in non-small-cell lung cancer
cell. PLoS One. 8:e653092013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sano D and Myers JN: Metastasis of
squamous cell carcinoma of the oral tongue. Cancer Metastasis Rev.
26:645–662. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Silverman S Jr: Demographics and
occurrence of oral and pharyngeal cancers. The outcomes, the
trends, the challenge. J Am Dent Assoc. 132:7S–11S. 2001.
View Article : Google Scholar
|
|
32
|
Chen X and Wang L: Integrating biological
knowledge with gene expression profiles for survival prediction of
cancer. J Comput Biol. 16:265–278. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lu J, Getz G, Miska EA, et al: MicroRNA
expression profiles classify human cancers. Nature. 435:834–838.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lovering RC, Camon EB, Blake JA and Diehl
AD: Access to immunology through the Gene Ontology. Immunology.
125:154–160. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Guo CJ, Pan Q, Li DG, Sun H and Liu BW:
miR-15b and miR-16 are implicated in activation of the rat hepatic
stellate cell: An essential role for apoptosis. J Hepatol.
50:766–778. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Omoto S and Fujii YR: Regulation of human
immunodeficiency virus 1 transcription by nef microRNA. J Gen
Virol. 86:751–755. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jiang L, Liu X, Chen Z, et al: MicroRNA-7
targets IGF1R (insulin-like growth factor 1 receptor) in tongue
squamous cell carcinoma cells. Biochem J. 432:199–205. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yao H, Wang H, Zhang Z, Jiang BH, Luo J
and Shi X: Sulforaphane inhibited expression of hypoxia-inducible
factor-1alpha in human tongue squamous cancer cells and prostate
cancer cells. Int J Cancer. 123:1255–1261. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Theocharis S, Klijanienko J, Giaginis C,
et al: Peroxisome proliferator-activated receptor-γ in mobile
tongue squamous cell carcinoma: associations with
clinicopathological parameters and patients survival. J Cancer Res
Clin Oncol. 137:251–259. 2011. View Article : Google Scholar
|
|
40
|
Bian L, Sun X, Jin K and He Y: Oral
cancer-associated fibroblasts inhibit heat-induced apoptosis in
Tca8113 cells through upregulated expression of Bcl-2 through the
Mig/CXCR3 axis. Oncol Rep. 28:2063–2068. 2012.PubMed/NCBI
|
|
41
|
Ozawa S, Kato Y, Komori R, Maehata Y,
Kubota E and Hata R: BRAK/CXCL14 expression suppresses tumor growth
in vivo in human oral carcinoma cells. Biochem Biophys Res Commun.
348:406–412. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lefort K, Brooks Y, Ostano P, et al: A
miR-34a-SIRT6 axis in the squamous cell differentiation network.
EMBO J. 32:2248–2263. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ries J, Vairaktaris E, Agaimy A, et al:
miR-186, miR-3651 and miR-494: potential biomarkers for oral
squamous cell carcinoma extracted from whole blood. Oncol Rep.
31:1429–1436. 2014.PubMed/NCBI
|
|
44
|
Zhang QH, Sun HM, Zheng RZ, et al:
Meta-analysis of microRNA-183 family expression in human cancer
studies comparing cancer tissues with noncancerous tissues. Gene.
527:26–32. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hassan MQ, Gordon JA, Beloti MM, et al: A
network connecting Runx2, SATB2 and the miR-23a~27a~24-2 cluster
regulates the osteoblast differentiation program. Proc Natl Acad
Sci USA. 107:19879–19884. 2010. View Article : Google Scholar
|
|
46
|
Carinci F, Lo Muzio L, Piattelli A, et al:
Potential markers of tongue tumor progression selected by cDNA
microarray. Int J Immunopathol Pharmacol. 18:513–524.
2005.PubMed/NCBI
|
|
47
|
Bijian K, Mlynarek AM, Balys RL, et al:
Serum proteomic approach for the identification of serum biomarkers
contributed by oral squamous cell carcinoma and host tissue
microenvironment. J Proteome Res. 8:2173–2185. 2009. View Article : Google Scholar : PubMed/NCBI
|