Introduction
Lithospermum erythrorhizon, a traditional
Chinese herbal medicine, has been used for the treatment of
inflammation, tumors and burns for thousands of years (1). Shikonin (SK) is the key active
ingredient isolated from the dried roots of Lithospermum
erythrorhizon, with a molecular weight of 288 g/mol (Fig. 1). As a naphthoquinone pigment, SK
exhibits multiple biological activities, including
anti-inflammatory, anti-microbial and anti-tumor effects, in
addition to antagonism of the human immunodeficiency virus and
wound healing-promoting properties (2,3).
Previous studies have demonstrated that SK induces apoptosis in
various types of tumor cell but has no effect on the majority of
normal cells (4,5), suggesting that it may provide a novel
anti-neoplastic therapeutic treatment strategy.
Various oncogenes and tumor suppressor genes are
involved in apoptosis, amongst which the tumor suppressor gene p53
is of particular significance (6).
p53 serves an important role in the prevention of tumor occurrence
and development via initiating cell cycle arrest and apoptosis in
damaged cells, in order to maintain genomic stability (7). The loss of wild-type p53 function may
lead to tumor development, and in humans, p53 is known to be
mutated or inactivated in at least 50% of tumors (8–10).
Certain studies have demonstrated that SK induces apoptosis in
Hela, colorectal carcinoma COLO 205 and malignant melanoma A375-S2
cells, accompanied by upregulated expression levels of p53 protein
(11–13). This suggested that SK-mediated
apoptosis involves p53 signaling.
However, in order to prolong their survival, tumor
cells have developed several mechanisms to inhibit apoptosis.
Previous studies have demonstrated that the phosphoinositide
3-kinase (PI3K)/Akt signaling pathway is persistently activated in
pre-malignant and malignant human bronchial epithelial cells in
addition to non-small cell lung cancer. This is suggested to be due
to activating mutations in ras (14–16).
In addition, multiple cytotoxic drugs have been reported to induce
apoptosis by inhibiting the PI3K/Akt pathway (17,18).
The casitas B-lineage lymphoma (Cbl) family of
ubiquitin ligases comprises adaptor proteins and E3 ligases that
serve positive and negative roles in several signaling pathways and
affect various cellular functions (19). Cbl proteins are known to
participate in the regulation of mitogen-activated protein kinases,
insulin signaling and PI3K/Akt signaling (19–21).
However, it remains elusive whether Cbl family members are involved
in SK-induced apoptosis.
Previous studies have further suggested that
reactive oxygen species (ROS) are involved in SK-induced apoptosis
in human SK-Hep-1 hepatoma cells and Bcr/Abl-positive chronic
myelogenous leukemia cells (22,23).
In the human oral squamous cell carcinoma Tca-8113 cell line,
induction of apoptosis by SK was demonstrated to be partly mediated
via inhibition of the nuclear factor-κB pathway (24). SK has also been identified to
downregulate estrogen receptor-α protein through a
proteasome-mediated pathway, and co-treatment with SK enhanced
sensitivity of breast cancer cells to endocrine therapy (25).
The present study aimed to investigate the
mechanisms of SK action in lung cancer A549 cells. The effects of
SK on the expression of p53 in A549 cells, in addition to the
association between Cbl and PI3K in SK-induced apoptosis of A549
cells, were examined. Additionally, the involvement of ROS in this
process was investigated. It was hypothesized that SK upregulates
the expression of Cbl proteins, which are involved in SK-induced
lung cancer cell apoptosis via negative regulation of PI3K/Akt
signaling.
Materials and methods
Reagents and antibodies
Rabbit polyclonal anti-B-cell lymphoma 2 (Bcl-2;
cat. no. sc-492), rabbit polyclonal anti-Bcl-2-associated X (Bax,
cat. no. sc-493), rabbit polyclonal anti-Cbl-b (cat. no. sc-1704)
and rabbit polyclonal anti-p53 (cat. no. sc-6243) antibodies were
purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
Rabbit monoclonal anti-phosphorylated (p)-Akt (Ser-473; cat. no.
3787s), rabbit polyclonal anti-Akt (cat. no. 9272), rabbit
monoclonal anti-c-Cbl (cat. no. C49H8) antibodies and the PI3K
inhibitor LY294002 were purchased from Cell Signaling Technology,
Inc. (Danvers, MA, USA). Rabbit polyclonal anti-β-actin (cat. no.
ab16039) was purchased from Abcam (Cambridge, UK). Goat anti-rabbit
horseradish peroxidase-conjugated secondary antibodies (cat. no.
A0208) were purchased from Beyotime Institute of Biotechnology
(Shanghai, China). SK (>98% pure) was purchased from the
National Institute for the Control of Pharmaceutical and Biological
Products (Beijing, China). SK was dissolved in dimethyl sulfoxide
(DMSO) at a stock concentration of 100 mM, aliquoted and stored at
−20°C. Fetal bovine serum (FBS) was purchased from Beijing Solarbio
Science & Technology Co., Ltd. (Beijing, China). MTT, DMSO,
propidium iodide (PI) and Hoechst 33342 were purchased from
Sigma-Aldrich (St. Louis, MO, USA). The annexin V-fluorescein
isothiocyanate and propidiumiodide (PI) double staining kit was
purchased from Beijing Biosea Biotechnology Co., Ltd. (Beijing,
China). Ribonuclease A (RNase A) and ROS assay kit were purchased
from Beyotime Institute of Biotechnology.
Cell culture
The human lung adenocarcinoma A549 cells were
purchased from the Department of Cell Biology, China Medical
University (Shenyang, China). A549 cells were cultured in RPMI 1640
medium (Hyclone, GE Healthcare, Little Chalfont, UK) containing 10%
FBS, 100 U/ml penicillin and 100 μg/ml streptomycin (Harbin
Pharmaceutical Group Co., Ltd., Harbin, China) at 37°C under an
atmosphere of 95% humidity and 5% CO2. Cells were
routinely subcultured every 2–3 days and cell samples used were all
in the logarithmic growth phase.
Cell viability assay
Cells were seeded at a density of 1×104
cells/well in 96-well plates and were incubated overnight.
Subsequently, various concentrations (0.312–10 μM) of SK
were added and further incubated for 24 and 48 h. Thereafter, 20
μl MTT solution (5 mg/ml) was added to each well and the
cells were incubated for an additional 4 h at 37°C. Following the
removal of culture medium, the cells were lysed in 150 μl
DMSO and the optical density was measured at 570 nm using a
microplate reader (Model 550; Bio-Rad Laboratories, Inc., Hercules,
CA, USA).
Cell cycle analysis
Phase distributions of the cell cycle and
hypodiploid DNA were determined by flow cytometry. A549 cells were
plated at a density of 1×106 cells/well in six-well
plates overnight and then treated with 1.5 and 1.8 μM SK for
24 h. Following this, the cells were collected and washed twice
with phosphate-buffered saline (PBS; Beyotime Institute of
Biotechnology). Subsequent to fixing in ice-cold 70% ethanol
(Shenyang Liaohe Chemical Plant, Shenyang, China) for 12 h, samples
were washed twice with PBS and then incubated with RNase A (20
μg/ml) and PI (10 μg/ml) for 30 min in the dark.
Finally, the samples were examined using a FACScan Flow Cytometer
and data were analyzed using CellQuest 3.2 software (BD
Biosciences, San Jose, CA, USA).
Cell apoptosis analysis
A549 cells were plated at a density of
1×106 cells/well in six well plates overnight and were
subsequently treated with 1.5 and 1.8 μM SK for 24 h. The
cells were collected following incubation. At the same time, A549
cells were plated in six well plates (1×106 cells/well)
overnight. The cells were pretreated with or without 10 nM Ps341
(Active Biochem, Maplewood, NJ, USA), an inhbitor of proteasome and
Cbl, for 1 h and subsequently treated with 1.8 μM SK for 24
h, prior to collection. The cells were washed twice with PBS and
incubated with 10 μl annexin V and 5 μl PI for 15 min
in the dark. The samples were evaluated by flow cytometry as
described above.
Fluorescence microscopy
A549 cells were treated with 1.5 and 1.8 μM
SK for 24 h. Subsequently, the cells were collected, washed twice
with PBS and fixed in a mixture of cold methanol and acetic acid
(3/1, v/v; Shenyang Liaohe Chemical Plant) prior to staining with
Hoechst 33342 (1 mg/ml) for 30 min at 37°C. Stained cells were
imaged using a fluorescence microscope (magnification, ×400;
Eclipse 90i, Nikon Corporation, Tokyo, Japan).
Western blot analysis
The expression of cellular proteins was evaluated by
western blotting. Following SK treatment for 4, 8, 12 and 24 h, and
pretreatment with or without 10 nM Ps341, cells were washed twice
with ice-cold PBS and the total proteins were solubilized and
extracted with lysis buffer (20 mM HEPES, pH 7.9; 20% glycerol; 200
mM KCl; 0.5 mM EDTA; 0.5% NP40; 0.5 mM DTT and 1% protease
inhibitor cocktail; Shanghai Jining Industrial Co., Ltd.). The
protein concentration was determined by the bicinchoninic acid
protein assay (Beyotime Institute of Biotechnology). Equal amounts
of protein (50 μg) from each sample were separated using 12%
SDS-PAGE (Beyotime Institute of Biotechnology, Shanghai, China).
Following electrophoresis, the proteins were electroblotted to
polyvinylidene difluoride membranes (Beyotime Institute of
Biotechnology). The membranes were blocked with 5% nonfat dry milk
dissolved in TBST (20 mM Tris-buffered saline, pH 7.5 containing 1
g/l Tween 20; Beyotime Institute of Biotechnology) at room
temperature and then independently incubated at 4°C overnight with
primary antibodies against Bcl-2 (1:300), Bax (1:300), p53
(1:1,000), Akt (1:1,000), p-Akt (1:1,000), c-Cbl (1:1,000) or Cbl-b
(1:250), and β-actin (1:1,000) was used as a control. Subsequent to
washing three times with TBST, the membranes were incubated with
goat anti-rabbit horseradish peroxidase-conjugated secondary
antibodies (1:800) at 37°C for 30 min, followed by a further three
washes with TBST. Protein bands were visualized using an enhanced
chemiluminescence reagent (Thermo Fisher Scientific, Waltham, MA,
USA).
Intracellular ROS measurement
A549 cells were treated with 1.5 and 1.8 μM
SK for 24 h and were then collected. Dichloro-dihydro-fluorescein
diacetate (DCFH-DA; 10 μM) was diluted at 1:1,000 with
serum-free RPMI-1640 medium (Gibco Life Technologies, Grand Island,
NY, USA). The cells were suspended in 1 ml diluted DCFH-DA, then
incubated in a 37°C cell incubator for 20 min with inversion every
3–5 min to maximize contact between the probe and the cells.
Following this, the cells were washed with serum-free cell culture
medium three times and the intracellular H2O2
concentration was determined using a FACScan flow cytometer (BD
Biosciences).
Statistical analysis
Values are expressed as the mean ± standard
deviation. Statistical correlation was evaluated by an analysis of
variance and Student’s t-test, where P<0.05 was considered to
indicate a statistically significant difference. These analyses
were performed using SPSS software, version 17.0 (SPSS, Inc.,
Chicago, IL, USA).
Results
SK induces growth inhibition of A549
cells
To determine whether SK inhibits proliferation of
lung cancer cells, A549 cells were treated with different
concentrations of SK (0.312–10 μM) for 24 and 48 h. A
significant dose-dependent and time-dependent reduction of
viability was observed (Fig. 2).
The 50% inhibitory concentration of cells (IC50) at 24
and 48 h was 1.75±0.26 and 1.25±0.17 μM, respectively.
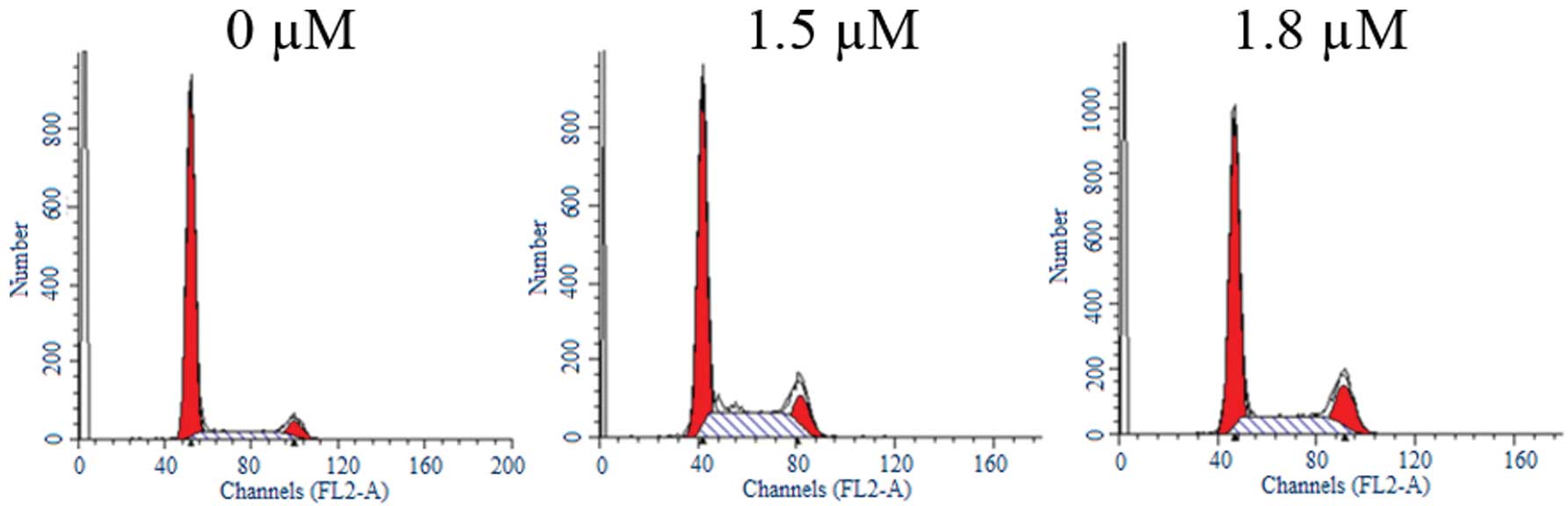
SK induces G2/M cell cycle
arrest in A549 cells
To determine whether SK inhibits cell cycle
progression in A549 cells, the cells were treated with different
concentrations of SK for 24 h. Flow cytometric analysis
demonstrated that treatment with SK induced significant
G2/M cell cycle arrest (Fig. 3). The percentages of cells in
G2/M phase were 5.42±1.06%, 12.65±2.24% and 17.27±3.27%
following treatment with 0, 1.5 and 1.8 μM SK, respectively
(Table I). Differences between
treated and untreated cells were statistically significant
(P<0.01).
 | Table ICell cycle distribution of A549 cells
incubated with SK for 24 h. |
Table I
Cell cycle distribution of A549 cells
incubated with SK for 24 h.
| SK (μM) | G1
(%) | S (%) | G2/M
(%) |
|---|
| 0 | 77.84±6.28 | 16.74±2.34 | 5.42±1.06 |
| 1.5 | 53.84±4.86 | 33.51±4.87 | 12.65±2.24 |
| 1.8 | 55.99±4.03 | 26.74±2.78 | 17.27±3.27 |
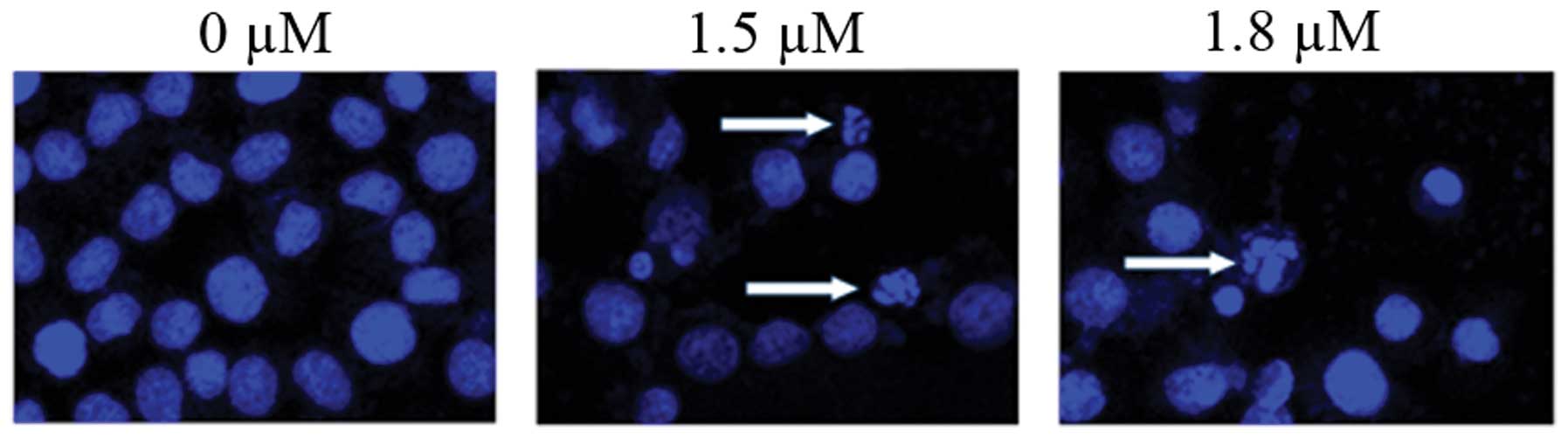
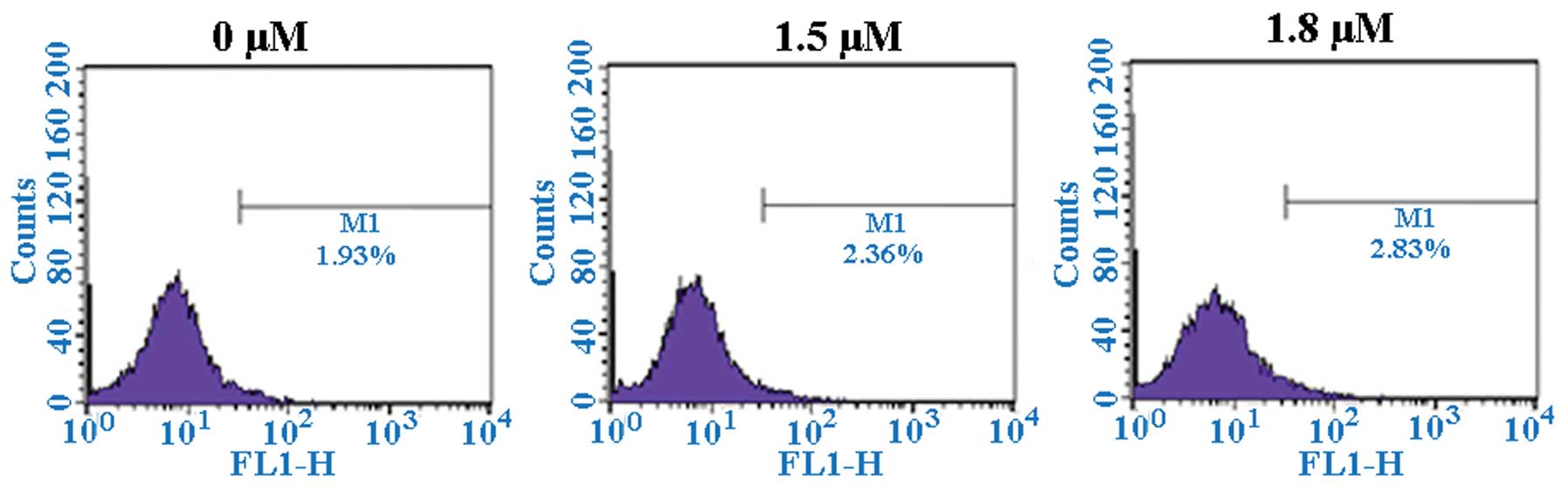
SK induces A549 cell apoptosis
To determine whether SK induces apoptosis in A549
cells, SK-induced cytotoxicity was investigated. Flow cytometric
analysis of annexin-V and PI demonstrated that SK induced
significant apoptosis in A549 cells. The percentages of
double-stained cells, indicative of apoptosis, were 17.20±2.26%
(P<0.01) and 21.98±2.26% (P<0.01) at 1.5 and 1.8 μM
SK, respectively, compared with 2.91±0.21% in the non-treated
control (Fig. 4). Consistent with
these results, confocal fluorescence microscopy also identified
clear morphological alterations typical of apoptosis, including
condensation of chromatin and nuclear fragmentations following
treatment of cells with SK (Fig.
5).
SK upregulates p53 and alters the
Bax/Bcl-2 ratio in favor of apoptosis
To determine the mechanism of SK-induced A549 cell
apoptosis, the expression levels of apoptosis-associated proteins
were investigated. Following incubation with either 1.5 or 1.8
μM SK for 24 h, the expression of the anti-apoptotic Bcl-2
protein was significantly reduced, whereas the expression of
pro-apoptotic Bax protein was significantly increased (Fig. 6). SK treatment significantly
increased the Bax/Bcl-2 ratio. It was hypothesized that p53 also
served an important role in SK-induced apoptosis. To investigate
this, the protein levels of p53 were examined in cells treated with
SK. SK was observed to increase the intracellular levels of p53 in
a dose-dependent manner (Fig.
6).
SK induces A549 cell apoptosis by
inhibiting PI3K/Akt signaling and potentiates LY294002-mediated
inhibition of proliferation and apoptosis
To further determine the mechanism of SK-induced
apoptosis in A549 cells, the levels of p-Akt and total Akt protein
following treatment with 1.8 μM SK for 4–24 h were measured.
It was observed that the expression levels of p-Akt protein started
to reduce at 4 h and reached a minimum at 24 h (Fig. 7). This suggested that SK induces
A549 cell apoptosis by inhibiting the PI3K/Akt signaling pathway.
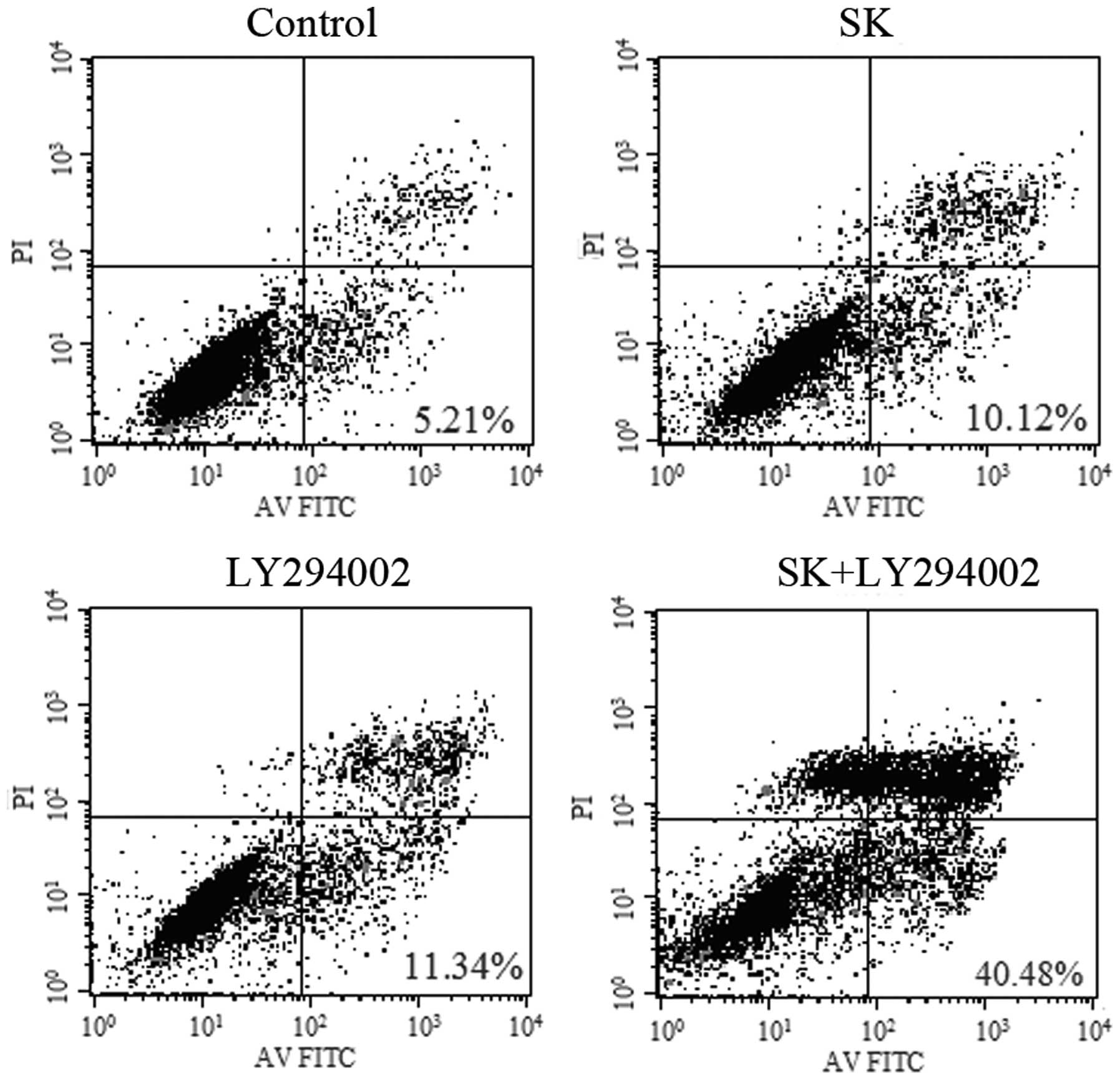
Additionally, 1-h pre-treatment of A549 cells with LY294002, a
specific inhibitor of PI3K, followed by the addition of 1 μM
SK (a sub-toxic dose), resulted in an increase in proliferation
inhibition. The proliferation inhibition of cells significantly
increased from 17.06±1.46 and 19.47±3.32% in cells treated with SK
alone and LY294002 alone, respectively, to 62.46±5.12% in cells
treated with both SK and LY294002 (P<0.01) (Fig. 8). In line with these observations,
in identically treated cells, apoptosis was identified to increase
from 10.12±2.48 and 11.34±3.38% to 40.68±6.46% (P<0.01)
(Fig. 9), while the expression
levels of p-Akt protein were significantly reduced (Fig. 10). These data suggested that
LY294002 and SK have a synergistic effect, leading to enhanced
inhibition of proliferation and induction of apoptosis in A549
cells.
Cbl-b and c-Cbl are involved in
SK-induced A549 cell apoptosis by negatively regulating PI3K/Akt
signaling
To investigate whether the inactivation of Akt was
associated with Cbl-b and c-Cbl, their protein expression levels
were assessed using western blotting (Fig. 11). In A549 cells treated with 1.8
μM SK for 4–24 h, the expression levels of the Cbl-b and
c-Cbl proteins were demonstrated to begin to increase at 4 h,
reaching a maximum at 24 h. The time taken for the upregulation of
Cbl-b and c-Cbl proteins was in accordance with that required for
the downregulation of the p-Akt protein. This indicated that the
inactivation of Akt was likely to be associated with the
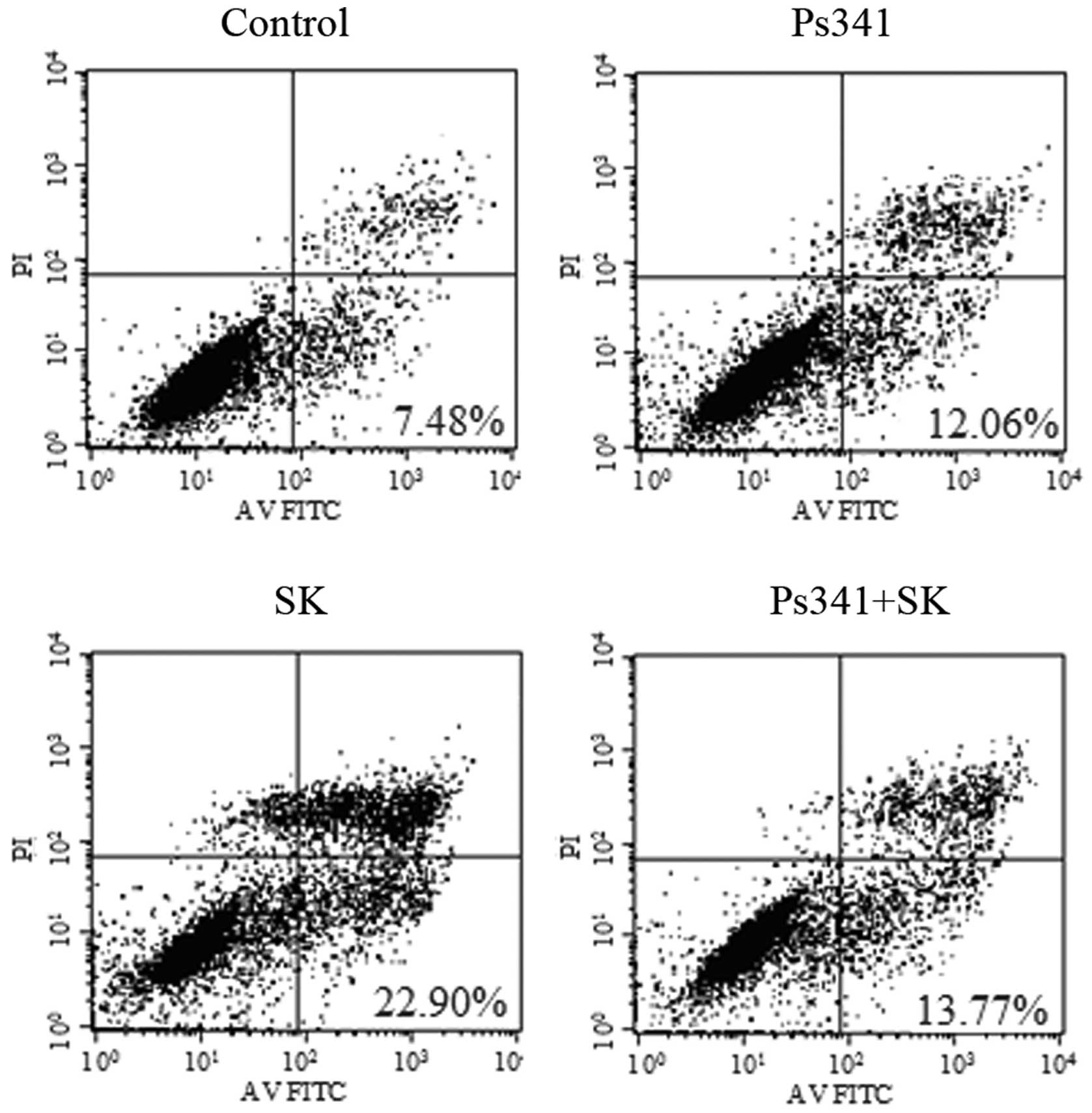
upregulation of Cbl-b and c-Cbl. To confirm this hypothesis, A549
cells were pre-treated with the proteasome inhibitor Ps341 and Cbl
for 1 h, then 1.8 μM SK was added for a further 24 h. Ps341
was observed to reverse SK-induced downregulation of p-Akt
(Fig. 12) and apoptosis of A549
cells (Fig. 13). These data
suggested that Cbl family members may be involved in SK-induced
A549 cell apoptosis via negatively regulating PI3K/Akt
signaling.
ROS is not involved in SK-induced
apoptosis
To address whether SK enhances ROS generation in
A549 cells, the intracellular H2O2
concentration was detected by DCFH-DA treatment and flow cytometric
analysis. As presented in Fig.
14, significant alterations in the ROS concentration were not
observed in cells treated with either 1.5 or 1.8 μM SK for
24 h. This result indicated that ROS does not participate in
SK-induced apoptosis in A549 cells.
Discussion
SK, one of the major naphthoquinone pigments
isolated from the traditional Chinese herb Lithospermum
erythrorhizon, has various biological functions. It is
associated with anti-microbial, anti-inflammatory and anti-tumor
activities, in addition to the promotion of wound healing. Previous
studies have demonstrated that SK is able to induce apoptosis in
liver cancer, osteosarcoma and prostate cancer cells while its
effect on normal cells is minimal. Thus use of SK may provide a
novel approach to the treatment of tumors (26–28).
The results of the present study indicated that SK inhibits A549
cell proliferation in a time- and dose-dependent manner. SK was
also observed to induce cell apoptosis and G2/M phase
cell cycle arrest. Several previous studies have investigated
SK-induced cell cycle arrest. SK has been demonstrated to induce
cell cycle arrest in the S phase of HepG2 cells
(26) and in the
G0/G1 phase of human bladder cancer cells
(4). Additionally, SK has been
identified to inhibit the cell cycle transition from G1
to S phase in Hela cells (29).
These results indicate that there is cell specificity in the effect
of SK on cell cycle arrest.
Apoptosis is the programmed cell death, which is
regulated by certain genes (30).
Apoptotic cells have unique morphology, characterized by cell
atrophy, loss of connection between cells and chromatin
condensation (31). Integration of
the apoptotic signal is predominantly mediated by Bcl-2 family
proteins and occurs at the mitochondrial level (32). The abnormal expression of Bcl-2
family members, including the pro-apoptotic proteins Bax, Bcl-2
homologous antagonist killer (Bak), Bcl-2-associated death promoter
(Bad), BH3 interacting-domain death agonist, Bcl-2-interacting
killer, Bcl-2-like protein 11, Harakiri and the anti-apoptotic
proteins Bcl-2, Bcl-2 extra large protein (Bcl-xL), Bcl-2-related
protein A1 and myeloid cell leukemia 1, which are represented by
Bax and Bcl-2, respectively, can aid tumor cells in evading
apoptosis and continuing to proliferate (33). The ratio of Bcl-2/Bax is suggested
to be a critical factor which determines the apoptosis threshold
(34). It has been reported that
in SK-induced apoptosis of Tca-8113 and HepG2 cells, the expression
of Bcl-2 protein is reduced (24,26),
while that of Bax is increased. Similarly, it has been demonstrated
that SK-induced COLO 205 cell apoptosis is accompanied by the
upregulation of Bad and downregulation of Bcl-2 and Bcl-xL, while
having no effect on Bax protein expression levels (29). The present study observed that SK
treatment resulted in a significant reduction in Bcl-2 protein
expression and a significant increase in Bax protein production in
A549 cells. It was therefore hypothesized that SK-mediated
inhibition of tumor cell growth is achieved by its ability to
modulate the ratio of Bcl-2/Bax.
The p53 gene is vital in preventing damaged or
otherwise abnormal cells from becoming malignant (12). Mutation factor induced-DNA damage
is able to promote rapid p53 gene product accumulation and the
subsequent induction of G1 phase cell cycle arrest
(35). p53 can also sequester
proliferating cell nuclear antigen, which inhibits the replication
of DNA and thereby allows time for DNA to be repaired. If the
damage cannot be repaired, p53 is able to induce apoptosis and
remove the mutant cells (36).
Additionally, p53 is involved in the mechanisms of apoptosis of
tumor cells induced by various drugs, including that mediated by
wogonin and arsenic trioxide (37,38).
The results of the present study demonstrated that the protein
expression levels of p53 were increased in SK-treated A549 cells,
and that the time required for p53 to increase coincided with that
for the increased ratio of Bax/Bcl-2. It has also been reported
that p53 can interact physically with Bcl-2 and Bcl-xL via its
DNA-binding domain, which leads to the isolation of these proteins
from their pro-apoptotic partners Bax/Bak. These proteins
subsequently form oligomers and increase the permeability of the
mitochondrial membrane, which leads to cytochrome C release into
the cytoplasm and cell apoptosis (39). Thus, it is suggested that SK first
upregulates the expression of the p53 protein, then subsequently
increases the ratio of Bax/Bcl-2 in order to induce A549 cell
apoptosis.
The PI3K/Akt signal transduction pathway is an
important intracellular cascade which promotes proliferation and
inhibits apoptosis by affecting the activity of downstream effector
molecules, including cell cycle regulatory proteins and
apoptosis-associated proteins (40). This pathway additionally serves
important roles in tumor cell proliferation, angiogenesis and
metastasis (41,42). The PI3K/Akt pathway is overactive
in certain types of cancer cell, while its inhibition may increase
the sensitivity of tumor cells to cytotoxic drugs and induce
apoptosis (43). In the present
study, treatment of A549 cells with 1.8 μM SK for 4–24 h
gradually reduced the protein expression of p-Akt, whereby
expression reached a minimum at 24 h. Typical apoptosis was
observed at 24 h, suggesting that SK first inhibited the PI3K/Akt
pathway, which in turn blocked its regulation of downstream
factors, resulting in the loss of PI3K/Akt pathway-mediated
anti-apoptotic function, and thus A549 cell apoptosis. This
indicates that inhibition of the PI3K/Akt pathway is likely to be
one of the mechanisms by which SK induces A549 cell apoptosis. The
association between SK and PI3K signaling in apoptotic induction of
A549 cells was further investigated using a specific inhibitor of
PI3K, LY294002, which inhibits the catalytic activity of the P110s
subunit. The results demonstrated that while 1 μM SK
resulted in a non-significant reduction in cell viability and no
greater than 11% cell apoptosis, treatment with SK plus LY294002
for 24 h resulted in a significant reduction in cell viability and
increased cell apoptosis to ~40% compared with treatment with SK or
LY294002 alone. These results indicated that LY294002 and SK have a
synergistic effect on the proliferation inhibition and apoptotic
induction of A549 cells.
The Cbl family members c-Cbl and Cbl-b, function as
E3 ubiquitin ligases that are able to interact with PI3K and
modulate its activity in various cell lines (16). By contrast, inhibition of Cbl-PI3K
interaction alters bone homeostasis, affecting osteoclast function
and osteoblast proliferation (37). Cbl is additionally involved in
arsenic trioxide-induced NB4 cell apoptosis and G2/M
arrest in gastric cancer cells via the inhibition of the PI3K/Akt
signaling pathway (15). The
results of the present study demonstrated that the protein
expression levels of c-Cbl and Cbl-b began to increase following
exposure of A549 cells to 1.8 μM SK for 4 h, and reached
maximal expression at 24 h. The kinetics associated with the
upregulation of c-Cbl and Cbl-b corresponded to Akt inhibition,
suggesting that SK is likely to promote PI3K ubiquitination via the
upregulation of c-Cbl and Cbl-b expression. This in turn inhibited
Akt activity and finally induced A549 cell apoptosis. Ps341, a Cbl
antagonist, was used to pre-treat A549 cells prior to the addition
of SK, and this was identified to reverse SK-induced cell apoptosis
in addition to the inhibition of p-Akt activity. These observations
suggested that c-Cbl and Cbl-b are involved in SK-induced A549 cell
apoptosis via the negative regulation of PI3K/Akt activity.
It has previously been demonstrated that treatment
with SK induces ROS generation, increases extracellular
signal-regulated kinase phosphorylation and reduces Bcl-2
expression. Additionally, pre-treatment with the antioxidant agent
N-acetyl cysteine has been observed to reverse SK-induced
ROS generation and significantly attenuate the cytotoxic effects of
SK on 143B osteosarcoma cells (27). SK has also been demonstrated to
induce human hepatocellular carcinoma Huh7 and BEL7402 cell
apoptosis through the ROS/Akt pathways (44). However, in the present study, ROS
was not identified to be involved in SK-induced A549 cell
apoptosis, suggesting that the involvement of ROS in SK-induced
apoptosis may be cell type-specific.
In conclusion, the present study demonstrated that
SK, a naturally occurring naphthoquinone, inhibits proliferation,
induces G2/M-phase arrest and induces apoptosis in A549
cells. SK induced A549 cell apoptosis by promoting the p53-mediated
increase of the Bax/Bcl-2 ratio in favor of apoptosis. The ligases
c-Cbl and Cbl-b additionally participated in SK-induced apoptosis
in A549 cells by negatively regulating the PI3K/Akt signaling
pathway.
References
|
1
|
Han W, Li L, Qiu S, Lu Q, Pan Q, Gu Y, Luo
J and Hu X: Shikonin circumvents cancer drug resistance by
induction of a necroptotic death. Mol Cancer Ther. 6:1641–1649.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen HM, Wang PH, Chen SS, Wen CC, Chen
YH, Yang WC and Yang NS: Shikonin induces immunogenic cell death in
tumor cells and enhances dendritic cell-based cancer vaccine.
Cancer Immunol Immunother. 61:1989–2002. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen X, Yang L, Zhang N, Turpin JA,
Buckheit RW, Osterling C, Oppenheim JJ and Howard OM: Shikonin, a
component of chinese herbal medicine, inhibits chemokine receptor
function and suppresses human immunodeficiency virus type 1.
Antimicrob Agents Chemother. 47:2810–2816. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yeh CC, Kuo HM, Li TM, Lin JP, Yu FS, Lu
HF, Chung JG and Yang JS: Shikonin-induced apoptosis involves
caspase-3 activity in a human bladder cancer cell line (T24). In
vivo. 21:1011–1019. 2007.
|
|
5
|
Wiench B, Eichhorn T, Paulsen M and
Efferth T: Shikonin directly targets mitochondria and causes
mitochondrial dysfunction in cancer cells. Evid Based Complement
Alternat Med. 2012:7260252012.PubMed/NCBI
|
|
6
|
Lago CU, Sung HJ, Ma W, Wang PY and Hwang
PM: p53, aerobic metabolism, and cancer. Antioxid Redox Signal.
15:1739–1748. 2011. View Article : Google Scholar :
|
|
7
|
Vogelstein B, Lane D and Levine AJ:
SUrfing the p53 network. Nature. 408:307–310. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Levine AJ: p53, the cellular gatekeeper
for growth and division. Cell. 88:323–331. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nigro JM, Baker SJ, Preisinger A, et al:
Mutations in the p53 gene occur in diverse human tumor types.
Nature. 342:705–708. 1989. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ryan KM, Phillips AC and Vousden KH:
Regulation and function of the p53 tumor suppressor protein. Curr
Opin Cell Biol. 13:332–337. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wu Z, Wu LJ, Tashiro S, Onodera S and
Ikejima T: Phosphorylated extracellular signal-regulated kinase
up-regulated p53 expression in shikonin-induced HeLa cell
apoptosis. Chin Med J. 118:671–677. 2005.PubMed/NCBI
|
|
12
|
Hsu PC, Huang YT, Tsai ML, Wang YJ, Lin JK
and Pan MH: Induction of apoptosis by shikonin through coordinative
modulation of the Bcl-2 family, p27 and p53, release of cytochrome
c and sequential activation of caspases in human colorectal
carcinoma cells. J Agric Food Chem. 52:6330–6337. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu Z, Wu L, Li L, Tashiro S, Onodera S and
Ikejima T: p53-mediated cell cycle arrest and apoptosis induced by
shikonin via a caspase-9-dependent mechanism in human malignant
melanoma A375-S2 cells. J Pharmacol Sci. 94:166–176. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tsao AS, McDonnell T, Lam S, Putnam JB,
Bekele N, Hong WK and Kurie JM: Increased phospho-AKT (Ser (473))
expression in bronchial dysplasia: implications for lung cancer
prevention studies. Cancer Epidemiol Biomarkers Prev. 12:660–664.
2003.PubMed/NCBI
|
|
15
|
Brognard J, Clark AS, Ni Y and Dennis PA:
Akt/protein kinase B is constitutively active in non-small cell
lung cancer cells and promotes cellular survival and resistance to
chemotherapy and radiation. Cancer Res. 61:3986–3997.
2001.PubMed/NCBI
|
|
16
|
Massion PP, Taflan PM, Shyr Y, et al:
Early involvement of the phosphatidylinositol 3-kinase/Akt pathway
in lung cancer progression. Am J Respir Crit Care Med.
170:1088–1094. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rasul A, Yu B, Khan M, Zhang K, Iqbal F,
Ma T and Yang H: Magnolol, a natural compound, induces apoptosis of
SGC-7901 human gastric adenocarcinoma cells via the mitochondrial
and PI3K/Akt signaling pathways. Int J Oncol. 40:1153–1161.
2012.
|
|
18
|
Jung KH, Choi MJ, Hong S, Lee H, Hong SW,
Zheng HM, Lee HS, Hong S and Hong SS: HS-116, a novel
phosphati-dylinositol 3-kinase inhibitor induces apoptosis and
suppresses angiogenesis of hepatocellular carcinoma through
inhibition of the PI3K/AKT/mTOR pathway. Cancer Lett. 316:187–195.
2012. View Article : Google Scholar
|
|
19
|
Swaminathan G and Tsygankov AY: The Cbl
family proteins: ringleaders in regulation of cell signaling. J
Cell Physiol. 209:21–43. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Naramura M, Band V and Band H:
Indispensable roles of mammalian Cbl family proteins as negative
regulators of protein tyrosine kinase signaling: Insights from in
vivo models. Commun Integr Biol. 4:159–162. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Thien CB, Dagger SA and Steer JH: c-Cbl
promotes T cell receptor-induced thymocyte apoptosis by activating
the phosphatidylinositol 3-kinase/Akt Pathway. J Biol Chem.
285:10969–10981. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mao X, Yu CR, Li WH and Li WX: Induction
of apoptosis by shikonin through a ROS/JNK-mediated process in
Bcr/Abl-positive chronic myelogenous leukemia (CMl) cells. Cell
Res. 18:879–888. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen CH, Chern CL, Lin CC, Lu FJ, Shih MK,
Hsieh PY and Liu TZ: Involvement of reactive oxygen species, but
not mitochondrial permeability transition in the apoptotic
induction of human SK-Hep-1 hepatoma cells by shikonin. Planta Med.
69:1119–1124. 2003. View Article : Google Scholar
|
|
24
|
Min R, Tong J, Wenjun Y, Wenhu D, Xiaojian
Z, Jiacai H, Jian Z, Wantao C and Chenping Z: Growth inhibition and
induction of apoptosis in human oral squamous cell carcinoma
Tca-8113 cell lines by Shikonin was partly through the inactivation
of NF-kappaB pathway. Phytother Res. 22:407–415. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yao Y and Zhou Q: A novel antiestrogen
agent Shikonin inhibits estrogen-dependent gene transcription in
human breast cancer cells. Breast Cancer Res Treat. 121:233–240.
2010. View Article : Google Scholar
|
|
26
|
Yingkun N, Lvsong Z and Huimin Y: Shikonin
inhibits the proliferation and induces the apoptosis of human HepG2
cells. Can J Physiol Pharmacol. 88:1138–1146. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chang IC, Huang YJ, Chiang TI, Yeh CW and
Hsu LS: Shikonin induces apoptosis through reactive oxygen
species/extracellular signal-regulated kinase pathway in
osteosarcoma cells. Biol Pharm Bull. 33:816–824. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang H, Zhou P, Huang H, et al: Shikonin
exerts antitumor activity via proteasome inhibition and cell death
induction in vitro and in vivo. Int I Cancer. 124:2450–2459. 2009.
View Article : Google Scholar
|
|
29
|
Wu Z, Wu LJ, Li LH, Tashiro S, Onodera S
and Ikejima T: Shikonin regulates HeLa cell death via caspase-3
activation and blockage of DNA synthesis. J Asian Nat Prod Res.
6:155–166. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Afford S and Randhawa S: Apoptosis. Mol
Pathol. 53:55–63. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lee DH, Kim C, Zhang L and Lee YJ: Role of
p53, PUMA, and Bax in wogonin-induced apoptosis in human cancer
cells. Biochem Pharmacol. 75:2020–2033. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Donovan M and Cotter TG: Control of
mitochondrial integrity by Bcl-2 family members and
caspase-independent cell death. Biochim Biophys Acta. 1644:133–147.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Anilkumar U and Prehn JH: Anti-apoptotic
BCL-2 family proteins in acute neural injury. Front Cell Neurosci.
8:2812014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang W, Guo Q, You Q, et al: Involvement
of bax/bcl-2 in wogonin-induced apoptosis of human hepatoma cell
line SMMC-7721. Anticancer Drugs. 17:797–805. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Taylor WR and Stark GR: Regulation of the
G2/M transition by p53. Oncogene. 20:1803–1815. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kastan MB and Kuerbitz SJ: Control of G1
arrest after DNA damage. Environ Health Perspect. 101:55–58.
1993.PubMed/NCBI
|
|
37
|
Lee DH, Rhee JG and Lee YJ: Reactive
oxygen species up-regulate p53 and Puma; a possible mechanism for
apoptosis during combined treatment with TRAIL and wogonin. Br J
Pharmacol. 157:1189–1202. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li Y, Qu X, Qu J, Zhang Y, Liu J, Teng Y,
Hu X, Hou K and Liu Y: Arsenic trioxide induces apoptosis and
G2/M phase arrest by inducing Cbl to inhibit PI3K/Akt
signaling and thereby regulate p53 activation. Cancer Lett.
284:208–215. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Scorrano L, Oakes SA, Opferman JT, Cheng
EH, Sorcinelli MD, Pozzan T and Korsmeyer SJ: BAX and Bak
regulation of endoplasmic reticulum Ca2+: a control point for
apoptosis. Science. 300:135–139. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Datta SR, Brunet A and Greenberg ME:
Cellular survival: A play in three Akts. Genes Dev. 13:2905–2927.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Brader S and Eccles SA: Phosphoinositide
3-kinase signalling pathways in tumor progression, invasion and
angiogenesis. Tumori. 90:2–8. 2004.PubMed/NCBI
|
|
42
|
Suthiphongchai T, Promyart P, Virochrut S,
Tohtong R and Wilairat P: Involvement of ERKI/2 in invasiveness and
metastatic development of rat prostatic Adenocarcinoma. Oncol Res.
13:253–259. 2003.
|
|
43
|
Tang YQ, Jaganath I, Manikam R and Sekaran
SD: Phyllanthus suppresses prostate cancer cell, PC-3,
proliferation and induces apoptosis through multiple signalling
pathways (MAPKs, PI3K/Akt, NFκB and Hypoxia). Evid Based Complement
Alternat Med. 2013:6095812013. View Article : Google Scholar
|
|
44
|
Gong K and Li W: Shikonin, a Chinese
plant-derived naphthoquinone, induces apoptosis in hepatocellular
carcinoma cells through reactive oxygen species: A potential new
treatment for hepatocellular carcinoma. Free Riadic Biol Med.
51:2259–2271. 2011. View Article : Google Scholar
|