Introduction
Myocardial ischemic postconditioning (IPostC)
treatment, applied at the onset of reperfusion, has been reported
to effectively ameliorate ischemia/reperfusion (I/R) injury of the
heart (1). Previous studies have
suggested that the protective effects of IPostC are predominantly
established via activation of the reperfusion injury salvage kinase
(RISK) pathway and the Janus kinase-signal transducer and activator
of transcription pathway by inhibiting mitochondrial permeability
transition pore opening (2,3).
Previous studies have also indicated that autophagy is involved in
the protective mechanisms of IPostC (4,5).
Autophagy is an intracellular degradation pathway,
in which long-lived or aggregated proteins and damaged organelles
are transported to lysosomes for destruction (6). In the heart, autophagy serves a
homeostatic role under physiological conditions, but abnormal
autophagy may result in cardiac dysfunction (7). A previous study demonstrated that
autophagy was upregulated during myocardial I/R and suggested that
it may be cardioprotective during ischemia (8). During myocardial ischemia and
reperfusion, autophagy is upregulated through activation of an
adenosine monophosphate-activated kinase (AMPK)- and Beclin
1-dependent mechanism (8). The
AMPK-mammalian target of rapamycin (mTOR) pathway and Beclin 1 are
core autophagy regulators, of which mTOR is additionally involved
in the RISK pathway. However, the involvement of autophagy during
the reperfusion period remains controversial (8–11).
The current study aimed to investigate the role of autophagy in
IPostC by comparing the myocardium autophagic flux and alterations
in the key autophagy regulators within a 24 h interval between
IPostC and I/R.
Materials and methods
Animals
A total of 192 male Sprague Dawley rats (SLAC
Laboratory Animals Ltd., Hunan, China) weighing 200–300 g were used
in the current study. The rats were housed in cages with ad
libitum access to food pellets and water, were maintained at
22°C, and were exposed to a 12 h light/dark cycle. The animal
protocol was approved by the Animal Care and Use Committee of
Xiangya Medical College, Central South University (Changsha,
China).
I/R modeling
Rats were anesthetized by intraperitoneal injection
of pentobarbital (60 mg/kg; Sigma-Aldrich, St. Louis, MO, USA) and
were placed on a heating pad (XR-YLS-20A; Xinruan Information
Technology, Co., Ltd., Shanghai, China) maintained at 36–38°C. An
electrocardiogram was recorded using a Power-Lab monitoring system
(BL-410 system; Chengdu Taimeng Science And Technology Co., Ltd.,
Chengdu, China). A left thoracotomy was subsequently performed to
expose the heart at the fourth intercostal space. Ischemia was
established by placing a snare on the top of the anterior
descending branch of the left coronary artery (LAD) using a 6-0
nylon suture (Cheng-He Microsurgical Instruments Factory, Ningbo,
China). Regional ischemia was confirmed by alterations in ST-T on
the electrocardiogram.
Western blot assay of autophagy
Rats were randomly divided into I/R, IPostC and
Sham-operation groups. Rats in the I/R group were subjected to 30
min ischemia to the heart followed by 1, 2, 3, 6, 12 and 24 h of
reperfusion (n=6 per time-point). Rats in the IPostC group were
subjected to 30 min ischemia to the heart prior to three cycles of
30-sec reperfusion/30-sec ischemia followed by 1, 2, 3, 6, 12 or 24
h reperfusion (n=6 per time-point). Rats in the Sham-operation
group (n=6) underwent the same procedure without occlusion of the
vessel. Rats were sacrificed by overdose of anesthesia
(pentobarbital, 180 mg/kg).
Hearts were excised and left ventricular (LV)
muscles were lysed in ice-cold radioimmunoprecipitation assay
buffer containing protease and phosphatase inhibitor cocktails (EMD
Millipore, Billerica, MA, USA). The concentration of the lysates
was measured using a Bicinchoninic Acid Protein Assay kit (Beyotime
Institute of Biotechnology, Beijing, China). Total protein (30
μg) was separated on 8–15% SDS polyacrylamide gels (Beyotime
Institute of Biotechnology) and transferred onto polyvinylidene
difluoride membranes (EMD Millipore). The membranes were blocked
with blocking buffer [5% bovine serum albumin (Wuhan Boster
Biological Technology, Ltd., Wuhan, China)-buffered saline
containing 0.05% Tween 20 (Sigma-Aldrich)] for 1 h at room
temperature. The blocked membranes were incubated with primary
antibodies overnight at 4°C. Subsequent to washing (Western Washing
Liquid; Beyotime Institute of Biotechnology), the membranes were
incubated with horseradish peroxidase-conjugated goat anti-rabbit
peroxidase-conjugated secondary antibody (1:1,000; cat. no. A0208;
Beyotime Institute of Biotechnology) for 1 h at room temperature.
Protein bands were visualized with the ECL Western Blotting
Detection kit (EMD Millipore), and the chemiluminescence signal was
detected using X-ray film (Kodak Digital Imaging Products Company
Ltd., Xiamen, China) with photographic cassettes (Beyotime
Institute of Biotechnology). Bands were scanned using AlphaView SA
version 3.3.0.0 software (ProteinSimple, San Jose, CA, USA). Rabbit
polyclonal Beclin 1 antibody (1:1,000; cat. no. 11306-1-AP) was
purchased from ProteinTech Group, Inc. (Chicago, IL, USA). Rabbit
polyclonal SQSTM/p62 (1:1,000; cat. no. 5114), rabbit polyclonal
LC3A/B (1:1,000; cat. no. 4108), rabbit monoclonal phospho-AMPKα
(Thr172) (1:1,000; cat. no. 2535), rabbit polyclonal
AMPKα (1:1,000; cat. no. 2532), rabbit monoclonal
phosphor-p70s6 kinase (Thr389) (1:1,000; cat.
no. 9234) and rabbit polyclonal P70S6 kinase (P70S6K) (1:1,000;
cat. no. 9202) antibodies were purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA). Rabbit polyclonal GAPDH
antibody (1:7,000; cat. no. ABS16) was purchased from EMD
Millipore.
Quantitative polymerase chain reaction
(qPCR)
The animal groupings and treatments in each group
were as described above. Samples were isolated from the LV muscle
of Sham-operated heart (Sham-operation group), the infarct LV
muscle of control hearts (I/R group) and the infarct LV muscle of
IPostC hearts (IPostC group). Total RNA was isolated using the
RNAiso Plus kit (Takara Biotechnology Co., Ltd., Dalian, China) and
cDNA was synthesized with 1 μg total RNA using gDNA Eraser
(Takara Biotechnology Co., Ltd.). A total of 2 μl cDNA
template was mixed with 18 μl 2X SYBR® Premix
EX Taq II mix (Takara Biotechnology Co., Ltd.) and this mixture
was subsequently subjected to qPCR at 95°C for 30 sec, followed by
40 cycles of 95°C for 5 sec and 60°C for 34 sec in the ABI 7500
Fast Real-Time PCR system (Applied Biosystems Life Technologies,
Foster City, CA, USA). GAPDH was amplified as an internal reference
gene. The following primers were used for amplification: i) Rat
microtubule associated protein 1 light chain 3 (LC3) b (LC3)
forward, 5′-AGC TCT GAA GGC AAC AGC AAC A-3′ and reverse, 5′-GCT
CCA TGC AGG TAG CAG GAA-3′; ii) rat SQSTM1 (p62) forward, 5′-TGT
GGT GGG AAC TCG CTA TAA GT-3′ and reverse, 5′-AAA GGG TTG GGA AAG
ATG AG-3′; iii) rat Beclin 1 forward, 5′-GAA ACT GGA CAC GAG CTT
CAA GA-3′ and reverse, 5′-ACC ATC CTG GCG AG-3′; iv) rat GAPDH
forward, 5′-GGC ACA GTC AAG GCT GAG AAT G-3′ and reverse, 5′-ATG
GTG GTG AAG ACG CCA GTA-3′ (Takara Biotechnology Co., Ltd.). The
comparative 2−ΔCt method (12) was utilized to analyze the relative
expression of genes.
Determination of myocardial infarct
size
Rats were randomly divided into the I/R, IPostC and
IPostC + chloroquine (CQ) groups. Rats in the I/R group were
subjected to 30 min ischemia to the heart followed by 3-h
reperfusion. Rats in the IPostC group were subjected to 30 min
ischemia to the heart prior to three cycles of 30-sec reperfusion
followed by 30-sec ischemia, and finally 3 h of reperfusion. Rats
in the IPostC + CQ group were pretreated with CQ (10 mg/kg,
intraperitoneally) 1 h prior to the induction of ischemia (13), and were subsequently subjected to
the same protocol as rats in the IPostC group (n=6–8 rats per
group). Rats were sacrificed by overdose of anesthesia
(pentobarbital, 180 mg/kg).
To measure the ischemic area at risk (AAR), the LAD
was re-occluded with the same suture. Evans blue dye (3%;
Sigma-Aldrich) was injected through the caudal vein. The heart was
excised and the LV tissues were cut into 1-mm cross-sections (Leica
CM1950 cryostat; Leica Microsystems GmbH, Nussloch, Germany). The
LV sections were incubated with a 1% triphenyltetrazolium chloride
solution (Sigma-Aldrich) at 37°C for 15 min. The infarct area
(pale), the AAR (not blue) and the total LV area from both sides of
each section were measured using Image-Pro Plus version 6.0 (Media
Cybernetics, Inc., Rockville, MD, USA).
Terminal deoxynucleotidyl transferase
mediated dUTP nick end labeling (TUNEL) staining
The hearts were rapidly excised following 3-h
reperfusion and were fixed in 4% para-formaldehyde (Sinopharm
Chemical Reagent Co., Ltd., Shanghai, China). The 10-μm
transverse cryostat sections were cut from routinely Tissue-Tek OCT
(Sakura Finetek, Inc., Torrance, CA, USA)-embedded heart tissues.
The slices were processed for TUNEL staining using a commercially
available in situ cell detection kit (Roche Diagnostics,
Indianapolis, IN, USA). Nuclei were stained with DAPI (Beyotime
Institute of Biotechnology). Apoptotic cell counts were expressed
as a percentage of the total number of nuclei.
Statistical analysis
Data were presented as the mean ± standard error of
the mean and were analyzed using SPSS version 17.0 (SPSS, Inc.,
Chicago, IL, USA). One-way analysis of variance was used for a
comparison between multiple groups and the Bonferroni post hoc test
was used for pairwise comparisons between groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
IPostC altered autophagic activity and
Beclin 1, AMPK-mTOR expression in a time-dependent manner
In the myocardium of rats treated with IPostC
compared with those of rats in the I/R group, the LC3-II/LC3-I
ratios were significantly lower at 1, 12 and 24 h but significantly
greater at 2, 3 and 6 h (P<0.05; Fig. 1A and B). The LC3-II protein levels
were significantly upregulated at 1 h but significantly
downregulated at the other time points (P<0.05; Fig. 1C). The p62 protein levels were
significantly greater at 1, 12 and 24 h but significantly reduced
at 2, 3 and 6 h (P<0.05; Fig. 1D
and E) Furthermore, the LC3b mRNA levels were observed to be
significantly lower at 1 and 24 h but significantly higher at 3 and
6 h (P<0.05; Fig. 2A), and the
p62 mRNA levels were also significantly lower at 1 and 24 h but
significantly higher at 2, 3 and 6 h (P<0.05; Fig 2B). Overall, IPostC was observed to
inhibit autophagic activity within the first hour, promote
autophagic activity from 2 to 6 h and reduce autophagic activity
from 12–24 h.
In the IPostC-treated myocardium, the Beclin 1 mRNA
levels were significantly lower at 1 h and significantly higher at
3, 6, 12 and 24 h, as compared with the I/R group (P<0.05;
Fig. 2C). The expression levels of
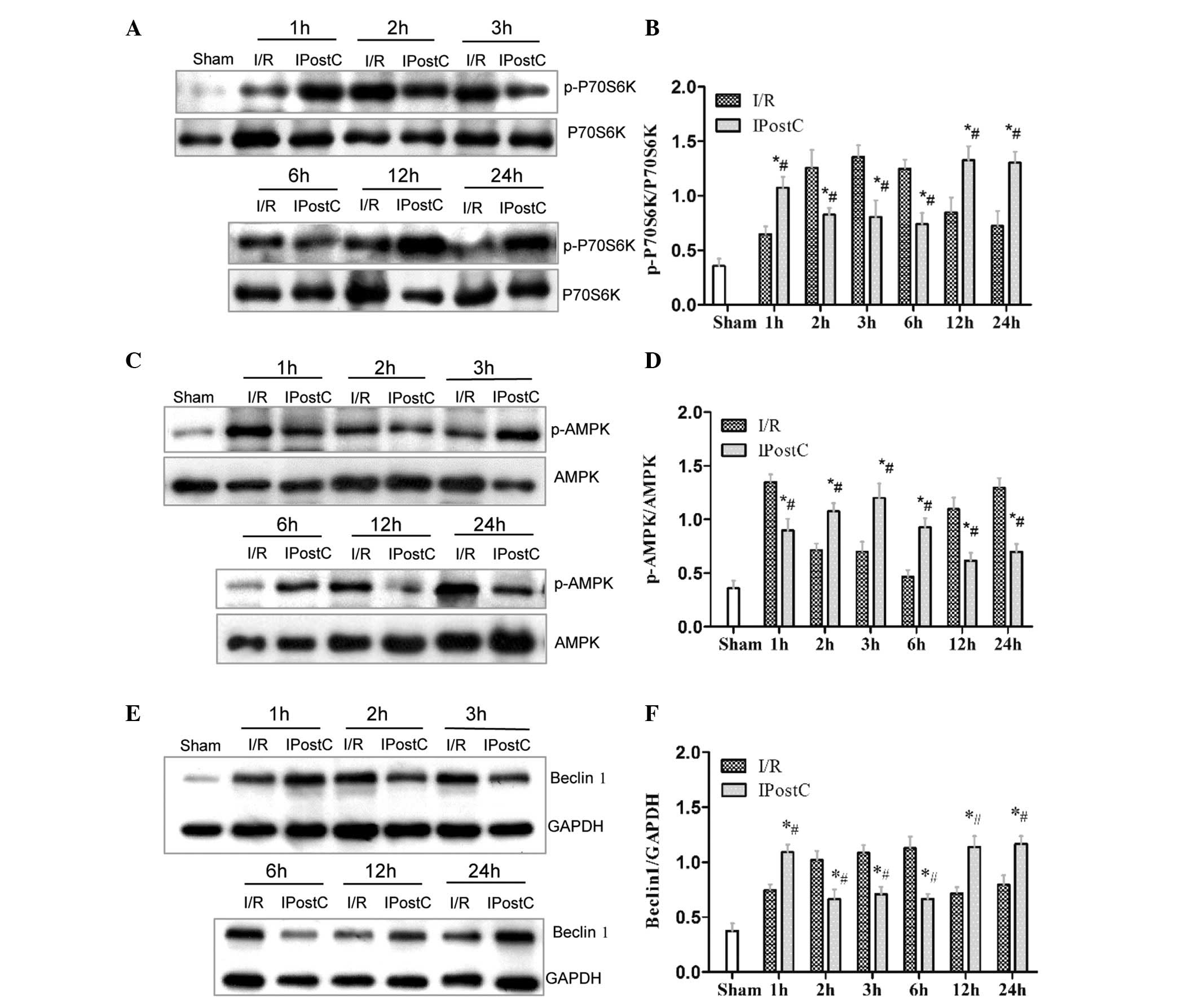
P70S6K were significantly increased following 1 h, reduced at 2, 3
and 6 h and increased again at 12 and 24 h (P<0.05; Fig. 3A and B). AMPK activity was
significantly inhibited at 1 h, increased at 2, 3 and 6 h and
reduced at 12 and 24 h (P<0.05; Fig. 3C and D). Beclin 1 protein levels
were significantly increased at 1 h, reduced at 2, 3 and 6 h,
followed by an increase again at 12 and 24 h (P<0.05; Fig. 3E and F).
Promotion of autophagic flux is
significant in conferring IPostC-mediated cardioprotection
A previous study demonstrated that the reperfused
myocardium exhibited impaired autophagosome clearance, which is
associated with cardiomyocyte death (14). The current study also observed
impaired autophagic flux in the reperfused myocardium following 2–6
h of reperfusion. Whether the relief of autophagy blocked by IPostC
in this period serves a significant role in its myocardial
protection was also investigated. The anti-malaria drug CQ is a
well-known autophagy inhibitor, which inhibits lysosomal
acidification and prevents autophagosome-lysosomal fusion (15). Rat hearts were pretreated with CQ 1
h prior to coronary artery occlusion. Following 3-h reperfusion,
the ischemic AAR was measured. The infarct volume in the hearts
subjected to I/R was 52% of the total LV area and this was reduced
to 26% (P<0.05) in hearts subjected to IPostC. By contrast, the
infarct volume in the IPostC + CQ rats was significantly increased
to 53% of the total LV area (P<0.05 vs. IPostC group; Fig. 4A and B). Furthermore, the
percentage of TUNEL-positive cells in the ischemic area was
additionally increased in the CQ-treated myocardium (17%) compared
with that of the IPostC-treated myocardium (7%) (Fig. 4C and D).
 | Figure 4Inhibition of autophagy reduces IPostC
cardioprotection. (A) Typical photographs of left ventricle tissue
stained by Evans blue and triphenyltetrazolium chloride dye. Rats
were subjected to 30 min of ischemia and 3 h of reperfusion with or
without IPostC and/or CQ treatment. Magnification, ×5. (B)
Quantitative analysis of infarct volume, n=6–8 per group. (C)
TUNEL-positive myocytes in the ischemic area. The number of
TUNEL-positive myocytes was expressed as a percentage of total
nuclei detected by DAPI staining, n=6 per group. (D) LV tissue
sections were subjected to TUNEL (red) and DAPI (blue) staining,
scale bar =50 μm. Error bars represent the mean ± standard
error of the mean; data were compared with a one-way analysis of
variance and post hoc Bonferroni test; *P<0.05 vs.
I/R group, #P<0.05 vs. IPostC group. IPostC, ischemic
postconditioning; TUNEL, terminal deoxynucleotidyl transferase
mediated dUTP nick end labeling; I/R, ischemia/reperfusion; CQ,
chloroquine; AAR, area at risk. |
Discussion
Autophagy has been demonstrated to be upregulated
during myocardial I/R and has been suggested to be cardioprotective
during ischemia. IPostC has been previously observed to function as
a protective mechanism against reperfusion injury (8). However, the role of autophagy in the
protective effects of IPostC and the associated signaling pathways
remain to be fully elucidated. The results of the current study
suggested that IPostC regulates autophagy in a time-dependent
manner through the regulation of Beclin 1 and AMPK-mTOR signaling
pathways.
Autophagy is a dynamic process and its activity is
predominantly reflected by autophagic flux, which is comprised of
autophagosome formation and autolysosome clearance (13). The conversion of LC3-I to LC3-II is
a marker of autophagic vesicle formation, however high ratios of
LC3-II/LC3-I may reflect an increase in autophagy, a reduction in
autophagic flux due to impaired fusion with lysosomes or
autolysosomal degradation. p62 is a protein adaptor, which is able
to bind ubiquitinated cargo designated for autophagic breakdown.
LC3-II, which is located on the inner autophagosomal membrane, is
degraded when the autophagosome fuses with the lysosome (16). Detecting the LC3-II/LC3-I ratio and
levels of p62 and LC3-II protein provides an improved
representation of autophagic flux (6,15).
p62 and LC3 are able to be transcriptionally regulated during
autophagy, which may influence the efficacy of p62 and LC3 levels
as indicators of autophagic substrates (15,17).
Despite these potential limitations, p62 and LC3b gene expression
levels were selected for detection in the myocardium during the
reperfusion period by qPCR. The RISK pathway predominantly includes
the phosphoinositide 3-kinase/protein kinase B (Akt)/mTOR pathway
and p42/p44 extracellular signal-regulated kinase pathway. These
two pathways converge at P70S6K. In mammalian cells,
phosphorylation of mTOR inhibits cell autophagy (17). Numerous studies demonstrate that
mTOR is a key factor for the regulation of autophagy in I/R
(18–22). P70S6K is an effector of mTOR, and
phosphorylated (p-)P70S6K levels are suggested to reflect the
function of mTOR (23). AMPK is
able to directly trigger autophagy or inhibit the suppressive
effect of mTOR (24). Beclin 1 is
a protein that controls the initiation of autophagy; however,
greater Beclin 1 levels additionally impede lysosomal fusion by
interacting with Rubicon or by directly downregulating
transcriptional components of the autophagy-lysosome machinery
(14,25). Therefore, Beclin 1 is a key
regulator, which exerts dual-directional regulation on autophagic
flux. For these reasons, the markers selected in the current study
are appropriate for the identification of autophagy and the
associated signaling.
During the first hour following treatment, reduced
gene expression of LC3b and a lower LC3-II/LC3-I ratio were
observed in the IPostC myocardium, which suggested a reduction in
autophagosome formation. Meanwhile, high p62 protein levels with
reduced p62 gene expression indicated that IPostC also inhibited
autophagy clearance. Therefore, it was suggested that IPostC
suppressed autophagic activity within the first hour following its
application. Consistent with a previous study (26), P70S6K was observed to be
significantly activated within 1 h following IPostC treatment,
suggesting that mTOR was also activated. In addition, a significant
reduction in AMPK activity was observed in the IPostC myocardium.
Thus, IPostC may negatively regulate autophagy via the activation
of mTOR. The expression of the Beclin 1 gene was reduced by IPostC
in this first hour, but the Beclin 1 protein level was upregulated
in the IPostC myocardium compared with the I/R group myocardium.
Upregulation of Beclin 1 has been previously identified to be
mediated by the high reactive oxygen species (ROS) content
resulting from reperfusion (14,27).
It is known that IPostC is able to reduce the production of ROS
(26), therefore a reduction in
the levels of ROS was hypothesized to contribute to the reduced
Beclin 1 expression during the first hour following IPostC
treatment. A previous study observed that Akt may enhance the
interaction among Beclin 1, 14-3-3 and vimentin, which inhibits the
role of Beclin 1 in autophagy (28). Therefore, activation of the RISK
pathway by IPostC may be caused by the accumulation of Beclin 1 in
the cytoplasm. The higher concentration of Beclin 1 may then
inhibit autophagic activity by impeding autophagy clearance or
downregulating the transcription of autophagy-associated genes
(14). Autophagy in the I/R
myocardium at 1 h appears to be over-activated when compared with
the myocardium in the IPostC group.
The excessive activation of autophagy may have
detrimental effects due to a disproportionate degeneration of
various key organelles and proteins (29). Thus, it is suggested that the
temporary inhibition of autophagy induced by IPostC produced a
protective effect on the myocardium.
The LC3-II/LC3-I ratio was observed to be higher in
the IPostC myocardium during 2–6 h post-ischemia and the gene
expression of LC3b was increased at 2 and 6 h, which suggested that
autophagosome formation was increased. In addition, reduced levels
of p62 protein expression together with significantly increased p62
gene expression levels indicates that IPostC also promotes
autophagy clearance. The LC3-II levels were reduced in the
IPostC-treated myocardium, which suggested that autophagy clearance
was promoted by IPostC, rather than the blockage of autolysosome
clearance induced by reperfusion. During this phase, P70S6K
activation was reduced and AMPK was activated again, an effect
which appeared to mediate autophagy activation in this period of
time. The Beclin 1 protein level was reduced and Beclin 1 gene
expression was increased in the IPostC myocardium compared with the
reperfused myocardium. The reduced Beclin 1 protein level may have
enhanced the autophagic flux during this time. Previous studies
have demonstrated that autophagic flux was blocked during
reperfusion (9,14). In the current study, it was
reported that reperfusion impaired autophagic flux between 2 and 6
h in I/R, and IPostC enhanced autophagic activity during this
period. It was further demonstrated that inhibition of autophagic
flux by CQ significantly increased myocardial infarct volume and
apoptosis, suggesting that relief of the impaired autophagic flux
by reperfusion is important in IPostC-induced cardioprotection.
During this period of time the reperfused myocardium exhibited
impaired autophagic flux, which may be an endogenous adaptation to
the overactivation of autophagy during the first hour; therefore,
blocking the autophagic flux temporarily produced a protective
effect on the myocardium. However, if this condition persists, it
is damaging to the myocardium due to the fact that the heart must
remove damaged proteins and organelles, particularly during the
reperfusion phase (30). A
previous study indicated that impaired autophagy was responsible
for cardiomyocyte death (14). The
enhanced autophagy induced by IPostC during this period produced a
protective effect on the myocardium.
At the 12 and 24 h time-points, the reduced
LC3-II/LC3-I ratio, LC3b and p62 gene expression levels together
with increased p62 protein expression levels suggested that
autophagic activity was reduced in the IPostC myocardium.
Simultaneously, P70S6K was observed to be activated once again.
Thus, mTOR may be involved in negative regulation of autophagy in
the IPostC myocardium. The Beclin 1 protein and gene expression
levels were all upregulated in the IPostC myocardium. In addition
to participating in autophagy, Beclin 1 is also involved in various
physiological processes, functioning as an important membrane
binding protein (31,32). Considering the reduced autophagic
activity, Beclin 1 may participate in additional physiological
processes in the IPostC myocardium at these time-points. Autophagic
activity was restored in the reperfused myocardium during the 12–24
h period, potentially as a result of the lower Beclin 1 levels
improving autophagic flux or due to the fact that myocardial cells
with autophagic dysfunction had already died. The autophagic
activity was reduced in the IPostC myocardium due to the fact that
the dysfunctional or damaged cellular components had been
eliminated and mTOR was re-activated, thus promoting the production
of proteins to repair the reperfused myocardium.
In conclusion, the current study highlighted the
crucial role of autophagy in the protective mechanism of IPostC via
regulation of the Beclin 1 and AMPK-mTOR signaling pathways.
However, further studies are required to confirm an association
between these signaling pathways and the regulation of autophagy.
Regulating autophagy in a time-dependent manner may be an
additional important end-effector of IPostC protection. Inhibition
or activation of autophagy during reperfusion may be harmful to the
myocardium, however regulation of autophagic activity in a
time-dependent manner may be an effective novel cardioprotective
treatment strategy.
References
|
1
|
Zhao ZQ and Vinten-Johansen J:
Postconditioning: reduction of reperfusion-induced injury.
Cardiovasc Res. 70:200–211. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hausenloy DJ: Signalling pathways in
ischaemic postconditioning. Thromb Haemost. 101:626–634.
2009.PubMed/NCBI
|
|
3
|
Ovize M, Baxter GF, Di Lisa F, et al
Working Group of Cellular Biology of Heart of European Society of
Cardiology: Postconditioning and protection from reperfusion
injury: where do we stand? Position paper from the Working Group of
Cellular Biology of the Heart of the European Society of
Cardiology. Cardiovasc Res. 87:406–423. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dosenko VE, Nagibin VS, Tumanovskaya LV,
Moibenko AA and Vaage J: Postconditioning prevents apoptotic
necrotic and autophagic cardiomyocyte cell death in culture. Fiziol
Zh. 51:12–17. 2005.PubMed/NCBI
|
|
5
|
Wei C, Li H, Han L, Zhang L and Yang X:
Activation of autophagy in ischemic postconditioning contributes to
cardio-protective effects against ischemia/reperfusion injury in
rat hearts. J Cardiovasc Pharmacol. 61:416–422. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Klionsky DJ, Abdalla FC, Abeliovich H, et
al: Guidelines for the use and interpretation of assays for
monitoring autophagy. Autophagy. 8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Matsui Y, Kyoi S, Takagi H, et al:
Molecular mechanisms and physiological significance of autophagy
during myocardial ischemia and reperfusion. Autophagy. 4:409–415.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Matsui Y, Takagi H, Qu X, et al: Distinct
roles of autophagy in the heart during ischemia and reperfusion:
roles of AMP-activated protein kinase and Beclin 1 in mediating
autophagy. Circ Res. 100:914–922. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hamacher-Brady A, Brady NR and Gottlieb
RA: Enhancing macroautophagy protects against ischemia/reperfusion
injury in cardiac myocytes. J Biol Chem. 281:29776–29787. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dosenko VE, Nagibin VS, Tumanovska LV and
Moibenko AA: Protective effect of autophagy in anoxia-reoxygenation
of isolated cardiomyocyte? Autophagy. 2:305–306. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Valentim L, Laurence KM, Townsend PA, et
al: Urocortin inhibits Beclin1-mediated autophagic cell death in
cardiac myocytes exposed to ischaemia/reperfusion injury. J Mol
Cell Cardiol. 40:846–852. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Iwai-Kanai E, Yuan H, Huang C, et al: A
method to measure cardiac autophagic flux in vivo. Autophagy.
4:322–329. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ma X, Liu H, Foyil SR, et al: Impaired
autophagosome clearance contributes to cardiomyocyte death in
ischemia/reperfusion injury. Circulation. 125:3170–3181. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Klionsky DJ, Abdalla FC, Abeliovich H, et
al: Guidelines for the use and interpretation of assays for
monitoring autophagy. Autophagy. 8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
He C and Klionsky DJ: Regulation
mechanisms and signaling pathways of autophagy. Annu Rev Genet.
43:67–93. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pattingre S, Espert L, Biard-Piechaczyk M
and Codogno P: Regulation of macroautophagy by mTOR and Beclin 1
complexes. Biochimie. 90:313–323. 2008. View Article : Google Scholar
|
|
19
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jung CH, Ro SH, Cao J, Otto NM and Kim DH:
mTOR regulation of autophagy. FEBS Lett. 584:1287–1295. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yu L, McPhee CK, Zheng L, et al:
Termination of autophagy and reformation of lysosomes regulated by
mTOR. Nature. 465:942–946. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Alers S, Löffler AS, Wesselborg S and
Srork B: Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy:
Cross talk, shortcuts, and feedbacks. Mol Cell Biol. 32:2–11. 2012.
View Article : Google Scholar :
|
|
23
|
Tsang CK, Qi H, Liu LF and Zheng XF:
Targeting mammalian target of rapamycin (mTOR) for health and
diseases. Drug Discov Today. 12:112–124. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mihaylova MM and Shaw RJ: The AMPK
signalling pathway coordinates cell growth, autophagy and
metabolism. Nat Cell Biol. 13:1016–1023. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhong Y, Wang QJ, Li X, et al: Distinct
regulation of autophagic activity by Atg14 L and Rubicon associated
with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol.
11:468–476. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Penna C, Mancardi D, Rastaldo R and
Pagliaro P: Cardioprotection: a radical view Free radicals in pre
and post-conditioning. Biochim Biophys Acta. 1787:781–793. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hariharan N, Zhai P and Sadoshima J:
Oxidative stress stimulates autophagic flux during
ischemia/reperfusion. Antioxid Redox Signal. 14:2179–2190. 2011.
View Article : Google Scholar :
|
|
28
|
Wang RC, Wei Y, An Z, et al: Akt-mediated
regulation of autophagy and tumorigenesis through Beclin 1
phosphorylation. Science. 338:956–959. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gustafsson ÅB and Gottlieb RA: Autophagy
in ischemic heart disease. Circ Res. 104:150–158. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gottlieb RA, Finley KD and Mentzer RM Jr:
Cardioprotection requires taking out the trash. Basic Res Cardiol.
104:169–180. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Huang W, Choi W, Hu W, et al: Crystal
structure and biochemical analyses reveal Beclin 1 as a novel
membrane binding protein. Cell Res. 22:473–489. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Funderburk SF, Wang QJ and Yue Z: The
Beclin 1-VPS34 complex-at the crossroads of autophagy and beyond.
Trends Cell Biol. 20:355–362. 2010. View Article : Google Scholar : PubMed/NCBI
|