Introduction
Acute and chronic exposure to particulate matter
(PM), particularly fine particles with aerodynamic diameters of
≤2.5 µm PM2.5, has been shown to increase the
number of hospital admissions for respiratory causes amongst the
general population (1). Evidence
obtained from environmental and epidemiological studies has
revealed a marked association between fine particulate air
pollution and multiple health issues, including respiratory
illnesses (for example, respiratory track inflammation, asthma,
acute bronchitis and lung cancer) as well as cardiovascular disease
mortality (2–4).
Increasing evidence supporting a link between
traffic-associated air pollution and the incidence of childhood
asthma has emerged; however, published estimates highlight the
variability observed between populations (5,6).
Asthma is a common worldwide respiratory symptom complex,
frequently involving airway inflammation, which results in
clinically significant physiological airway dysfunction. Asthma
pathogenesis is complex, and multiple factors have roles in the
development of the disease (7).
Autophagy describes an evolutionarily conserved and
tightly regulated lysosomal pathway, responsible for the
degradation of macromolecules, including proteins, glycogen,
lipids, nucleotides and organelles, via several complex pathways
(8). Recently, this process has
been demonstrated to be involved in numerous biological processes,
including host defense, cell survival and cell death. The detection
of autophagosomes in fibroblasts and epithelial cells in tissues
from patients with asthma has revealed a potential link between
autophagy and asthma pathogenesis (9). Numerous stress signals are able to
induce autophagy, including p53, phosphatidylinositol 3-kinase
(PI3K)/mammalian target of rapamycin (mTOR) and adenosine
monophosphate-activated protein kinase (AMPK) (10,11).
The PI3K/Akt/mTOR signaling pathway is required for the regulation
of autophagic flux. The mitogen activated protein kinase (MAPK)
pathway mediates the transmission of signals between receptors and
the nucleus via multiple intermediate proteins, including Ras, Raf,
extracellular signal-regulated kinase (ERK) and MAPK/ERK (MEK).
Akhtar et al (12) reported
that the induction of autophagy via pathways involving ERK1/2, p38
MAPK and Akt enhanced cardiac cell survival following
ischemia-reperfusion injury.
A previous study by our group revealed that p53
mediated PM-induced alveolar epithelial cell mitochondria-regulated
apoptosis (13). It was also
confirmed that the Akt/mTOR and c-Jun N-terminal kinase (JNK)
signaling pathways were involved in chrysotile asbestos-induced
autophagy in human lung epithelial cells (A549) (14). In the present study, the effect of
PM2.5 on the induction of autophagy in vitro and
the potential underlying mechanisms were evaluated.
Materials and methods
PM2.5 sampling and
composition
The PM2.5 used in the present study was
obtained from Zhanjiang, China. The PM2.5 was collected
using the QJS-100 multi-level flow particulate matter cutter
(Jinzhoulicheng, Jinzhou, China), at a constant aspiration flow
rate (100 l/min) over a period of 48 h. The sample containing the
PM2.5 fiber was placed in ultrapure water and subjected
to ultrasonic oscillations (Xingzhi Biotechnology Co., Ltd.,
Ningbo, China) for 15 min to elude the particulate matter,
vacuum-freeze dried (Xingzhi Biotechnology Co., Ltd.,) for 24 h and
formulated into a stock solution by the addition of
phosphate-buffered saline (PBS), followed by auto-claving. The
PM2.5 suspensions were vortexed and stored at 4°C.
PM2.5 filter samples were analyzed according to a
previous study (15), the results
of which are exhibited in Table
I.
 | Table IConcentrations of 15 types of PAHs,
transition metals and common metals identified in PM from
Zhanjiang, China. |
Table I
Concentrations of 15 types of PAHs,
transition metals and common metals identified in PM from
Zhanjiang, China.
| PM Content | Concentration
(pg/µg) |
|---|
| PAHs | |
| Napthalene | 23.11 |
| Acenapthylene | 25.00 |
| Fluorene | 42.22 |
| Phenanthrene | 26.89 |
| Anthracene | 20.33 |
| Fluoranthene | 28.00 |
| Perylene | 36.11 |
|
1,2-benza(a)anthracene | 23.33 |
| Chrysene | 27.78 |
|
Benzo(b)fluoranthene | 61.56 |
|
Benzo(k)fluoranthene | 35.00 |
|
Benzo(a)pyrene | 17.22 |
|
Ideno(1,2,3-cd)pyrene | 24.44 |
|
Dibenzo(ah)anthracene | 30.56 |
| Benzopyrene | 11.33 |
| Total PAHs | 459.56 |
| Metals | Concentration
(ng/µg) |
| Na | 12.44 |
| K | 19.44 |
| Mg | 4.67 |
| Ca | 3.89 |
| Fe | 3.00 |
| Cu | <0.1 |
| Cr | 0.11 |
| Co | <0.1 |
| V | <0.1 |
| Ni | 0.72 |
| Cd | <0.1 |
| As | <0.1 |
Reagents and antibodies
Dulbecco’s modified Eagle’s medium (DMEM),
penicillin, streptomycin and fetal bovine serum (FBS) were
purchased from Gibco-BRL Life Technologies (Gaithersburg, MD, USA).
Rapamycin, dimethyl sulfoxide (DMSO) and trypsin-EDTA were obtained
from Sigma-Aldrich (St. Louis, MO, USA). Rapamycin was dissolved in
DMSO and the final concentration of DMSO in the culture medium did
not exceed 0.2%. The primary antibodies used in western blotting
and immunofluorescent analysis are exhibited in Table II. All secondary antibodies were
obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
3-MA, dorsomorphin, pifithrin-α (PFT-α) and tetramethylthiourea
(TMTU) were obtained from Sigma-Aldrich. LY294002, U0126, SP600125
and SB203580 were purchased from Calbiochem® (Merck
Millipore KGaA, Darmstadt, Germany). pBABE-mCherry-enhanced-green
fluorescent protein (eGFP)-microtubule-associated protein 1 light
chain 3 (LC3) was purchased from Biovector Science Lab, Inc.
(Beijing, China). SDS sample buffer, protease inhibitors, nonfat
dry milk, Tris-buffered saline (TBS) and Tween 20 were purchased
from Sangon Biotech (Shanghai) Co., Ltd. (Shanghai, China).
Lipofectamine® 2000 was obtained from Invitrogen Life
Technologies (Carlsbad, CA, USA). Glutaraldehyde,
parafor-maldehyde, cacodylate buffer, OsO4, ethanol,
epoxy resin, lead citrate and uranyl acetate were purchased from
Guangzhou Chemical Reagent Factory (Guangzhou, China).
| Table IIPrimary antibodies used in the
present study. |
Table II
Primary antibodies used in the
present study.
| Antibody | Source | Clonality | Manufacturer (cat.
no.) | Concentration |
|---|
| Anti-AKT | Rabbit | Polyclonal | Cell Signaling
Technology, Inc. (#9272) | 1:300 (WB) |
| Anti-p-AKT | Rabbit | Monoclonal | Cell Signaling
Technology, Inc. (#4058) | 1:300 (WB) |
| Anti-AMPK | Rabbit | Monoclonal | Cell Signaling
Technology, Inc. (#5831) | 1:300 (WB) |
| Anti-p-AMPK | Rabbit | Polyclonal | Cell Signaling
Technology, Inc. (#2531) | 1:300 (WB) |
| Anti-ERK | Rabbit | Monoclonal | Cell Signaling
Technology, Inc. (#4695) | 1:300 (WB) |
| Anti-p-ERK | Rabbit | Monoclonal | Cell Signaling
Technology, Inc. (#4377) | 1:300 (WB) |
| Anti-JNK | Rabbit | Monoclonal | Cell Signaling
Technology, Inc. (#9258) | 1:300 (WB) |
| Anti-p-JNK | Rabbit | Monoclonal | Cell Signaling
Technology, Inc. (#4668) | 1:300 (WB) |
| Anti-p38 | Rabbit | Polyclonal | Cell Signaling
Technology, Inc. (#9212) | 1:300 (WB) |
| Anti-p-p38 | Rabbit | Monoclonal | Cell Signaling
Technology, Inc. (#4511) | 1:300 (WB) |
| Anti-p53 | Rabbit | Monoclonal | Cell Signaling
Technology, Inc. (#2527s) | 1:300 (WB) |
| Anti-p-p53 | Rabbit | Polyclonal | Cell Signaling
Technology, Inc. (#9284) | 1:300 (WB) |
| Anti-β-actin | Rabbit | Polyclonal | Santa Cruz
Biotechnology, Inc. (sc-1616) | 1:500 (WB) |
| Anti-mTOR | Rabbit | Polyclonal | Cell Signaling
Technology, Inc. (#2972) | 1:300 (WB) |
| Anti-p-mTOR | Rabbit | Monoclonal | Cell Signaling
Technology, Inc. (#5536) | 1:300 (WB) |
| Anti-LC3 | Rabbit | Polyclonal | Cell Signaling
Technology, Inc. (#4108) | 1:300(WB) |
| | | | 1:100(IF) |
Cell culture and treatments
Beas-2B human bronchial epithelial cells were
purchased from the American Type Culture Collection (Manassas, VA,
USA). Beas-2B cells were cultured in DMEM supplemented with 10%
heat-inactivated FBS, penicillin (50 U/ml) and streptomycin (50
U/ml). The cells were incubated at 37°C with humidified 5%
CO2. Cells were seeded at a concentration of
1.5×105 cells/well in six-well plates. Following 24 h of
culture, cells were treated with PM2.5 and rapamycin at
concentrations of 100 µg/ml in 10% FBS supplemented medium
for the indicated time-periods. For the analysis of PI3K and MAPK
signaling pathway inhibition, Beas-2B cells were pre-treated with
inhibitors 3-MA (Sigma-Aldrich; 10 mM), LY294002 (Merck Millipore;
10 nM), U0126 (Merck Millipore; 5 µM), SP600125 (Merck
Millipore; 50 µM) and SB203580 (Merck Millipore; 10
µM) for 2 h at 37°C, prior to treatment with 100
µg/ml PM2.5 for 24 h at 37°C in the presence of
the inhibitors. The inhibitors were dissolved in DMSO and the final
DMSO concentration in the culture medium was ≤0.2%.
Transmission electron microscopy
(TEM)
The Beas-2B cells were cultured on plates and
treated with PM2.5 for 24 h prior to fixing with 3%
glutaraldehyde and 2% paraformaldehyde (PFA) in 0.1 mol/l
cacodylate buffer (pH 7.3) for 1 h. Following fixation, the samples
were postfixed in 1% OsO4 in identical buffer for 1 h,
serially dehydrated with ethanol and embedded in epoxy resin.
Subsequently, sections (70 nm) were cut on a Leica Ultra-CUT
(Ultra-Microtome; Leica Microsystems GmbH, Wetzlar, Germany) and
contrasted with 0.1% lead citrate and 8% uranyl acetate in 50%
ethanol. Subsequently, the ultrathin sections were evaluated under
a transmission electron microscope (JEM-1400; JEOL Ltd., Tokyo,
Japan) operated at 120 kV, and images were captured using a
Megaview III CCD camera (Soft Imaging System, Lakewood, CO,
USA).
Western blot analysis
Cells were washed twice in PBS and protein extracts
were obtained by solubilizing the cells in SDS sample buffer
supplemented with protease inhibitors. Soluble proteins were
isolated from the untreated or treated Beas-2B cells for western
blot analysis as described previously (16). Equal amounts of protein (20
µg) from each sample were separated by electrophoresis on
10% SDS-PAGE, transferred to polyvinylidene difluoride membranes
(Merck Millipore KGaA) and blocked with 5% nonfat dry milk in 1X
TBS plus 0.1% Tween 20 at room temperature for 1 h. The membranes
were subsequently incubated with primary antibodies diluted in 5%
nonfat dry milk in 1X TBS plus 0.1% Tween 20 overnight at 4°C. The
primary antibody against actin (diluted 1:500) was purchased from
Santa Cruz Biotechnology, Inc. (Danvers, MA, USA). Following
incubation with primary antibodies (Table II), the membranes were washed and
incubated with horseradish peroxidase-conjugated anti-mouse
(sc-2025) or anti-rabbit (sc-2027) secondary antibodies for 1 h at
room temperature. The bound antibodies were detected using an
Enhanced Chemiluminescence Western Blotting system (Amersham
Biosciences Corp., Piscataway, NJ, USA).
Immunofluorescent microscopy of cells
exhibiting LC3-positive vesicles
For indirect immunofluorescent staining, cells were
fixed with 4% PFA for 20 min, incubated with blocking buffer
(comprised of 3% bovine serum albumin and 0.01% saponin) for 45
min. In order to analyze the autophagic flux, Beas-2B cells were
transfected with mCherry-eGFP-LC3-expressing plasmids. The cells
were pooled and seeded in chamber slides at a density of
2×104. The level of autophagic flux was determined via
examination of the punctate pattern of eGFP and mCherry expression
by counting the puncta per cell. Fluorescent images were captured
with a confocal microscope (TCS SP5 II; Leica Microsystems GmbH)
and analyzed using Cell M software (TCS SP5 II).
Flow cytometric analysis of
apoptosis
Apoptosis analysis was performed using flow
cytometry with propidium iodide (PI) staining. Cells were seeded at
a density of 5×105 cells/well in six-well plates and
following 24 h of culture, were treated with 100 µg/ml
PM2.5 for a further 24 h at 37°C. Following
PM2.5 exposure, cells were harvested and subjected to
centrifugation at 400 × g for 5 min at 4°C. The cells were fixed by
incubation with cold 75% ethanol for 24 h and subsequently stained
with PI solution, which was comprised of 45 mg/ml PI, 10 mg/ml
RNase A and 0.1% Triton X-100. Following incubation at 4°C for 1 h
in the dark, the fluorescence-activated cells were evaluated using
a FACScan flow cytometer (BD Biosciences, Franklin Lakes, NJ,
USA).
Statistical analysis
Values are presented as the mean ± standard
deviation. Statistical differences among experimental groups were
evaluated using one-way analysis of variance with repeated
measures. P<0.05 was considered to indicate a statistically
significant difference between values. The statistical analysis
software package SPSS 11.0 for Windows (SPSS Inc., Chicago, IL,
USA) was used to conduct the statistical analyses.
Results
PM2.5 induces the expression
of morphological and biochemical markers of autophagy in Beas-2B
cells
Treatment of human Beas-2B cells with
PM2.5, induced the expression of morphological and
biochemical markers of autophagy. Initially, the effects of
PM2.5 were analyzed by incubating Beas-2B cells with
three concentrations of PM2.5 (25, 50 and 100
µg/ml), or with PBS as the negative control for 24 h
(Fig. 1A). Exposure to
PM2.5 for 24 h resulted in an up to two-fold,
dose-dependent increase in LC3I to LC3II processing, with an
apparent maximum at 50 µg/ml. Exposure of Beas-2B cells to
100 µg/ml PM2.5 for 12, 24 or 48 h induced a
time-dependent increase in LC3I to LC3II processing, which
commenced at 12 h and continued to increase up until 24 h of
exposure. Whether there was efficient autophagic flux upon exposure
to PM2.5 was subsequently examined. Beas-2B cells were
transfected with the tandem-tagged, fluorescent reporter plasmid
mCherry-eGFP-LC3II (Fig. 1B). An
increase in autophagic flux is indicated by enhanced expression of
yellow and red puncta within the cells, whereas blocking of
autophagic flux is indicated by an increase in yellow puncta,
without an accompanying increase in the number of red puncta in the
cells (17). PM2.5
treatment of Beas-2B cells revealed significant accumulation of
yellow and red foci, indicative of an enhanced population of
immature autophagosomes. These data suggested that PM2.5
induced autophagy and enhanced autophagic flux in Beas-2B
cells.
 | Figure 1Autophagy is induced in Beas-2B cells
following exposure to PM2.5. (A) Beas-2B human bronchial
epithelial cells were treated with the indicated concentrations of
PM2.5 for 24 h or with 100 µg/ml PM2.5
for the indicated time-periods. Cell lysates were subjected to
immunoblot analysis for detection of LC3 levels and β-actin was
used as loading control. Quantification of the results are
presented as the amount of LC3II normalized against LC3I. (B)
Beas-2B cells were transfected with mCherry-eGFP-LC3 and treated
with 100 µg/ml PM2.5 for 24 h. Beas-2B cells were
treated with phosphate-buffered saline and rapamycin as negative
and positive controls, respectively. Cells were examined by
fluorescent microscopy, and representative cells were selected and
photographed. (C) PM2.5 induced ultrastructural features
of autophagy. Beas-2B cells were treated with 100 µg/ml
PM2.5 for 24 h and processed for electron microscopy.
Note the double membrane structure of the autophagic vacuoles.
Degrading autophagic vacuoles (AVds) are indicated. Scale bar, 500
nm. Values are expressed as the mean ± standard deviation of three
independent experiments. *P<0.05,
**P<0.01 and ***P<0.001 vs. control.
All above experiments were repeated three times. PM2.5,
particle matter 2.5; LC3, microtubule-associated protein 1 light
chain 3; NC, normal control; N, nucleus; Mit, mitochondria; eGFP,
enhanced green fluorescent protein; AV, autophagic vesicles. |
Morphological indices of autophagy were also
evaluated in PM2.5-treated Beas-2B cells. Exposure to
PM2.5 for 24 h resulted in an increase in the formation
of immature and degradative autophagic vesicles in Beas-2B cells,
as detected by electron microscopy (Fig. 1C). Quantification of electron
micrographs revealed an approximately three-fold increase in the
number of autophagy-positive cells in PM2.5-exposed
Beas-2B cells compared with that of PM2.5-untreated
cells. These data further supported the hypothesis that
PM2.5 induces autophagy in Beas-2B cells.
The AMPK signaling pathway is activated,
but not required, for PM2.5-induced autophagy
Previous studies have demonstrated that AMPK
functions as an upstream kinase for mTOR, and that AMPK activation
downregulates mTOR signaling (18). It was therefore hypothesized that
AMPK activation may provide the upstream kinase involved in
autophagy activation. In order to investigate the effect of the
AMPK signaling pathway, the expression and phosphorylation of AMPK
(phosphorylation sites at Thr-172) was examined in Beas-2B cells.
As shown in Fig. 2,
PM2.5 enhanced the phosphorylation of AMPK following 24
h of incubation. Immunoblot analysis revealed that this enhanced
phosphorylation did not occur in tandem with an increase in total
AMPK expression (Fig. 2A).
Dorsomorphin, a specific AMPK inhibitor, was used to block AMPK
phosphorylation. Western blot analysis demonstrated that
dorsomorphin effectively blocked AMPK activation in Beas-2B cells;
however, PM2.5-induced autophagy was not significantly
decreased by dorsomorphin treatment as revealed by LC3II expression
analysis (Fig. 2). These results
indicated that AMPK is activated by PM2.5 exposure for
24 h; however, this activation is not required for
PM2.5-induced autophagy.
The Akt/mTOR signaling pathway is
involved in PM2.5-induced autophagy
The PI3K/Akt/Raptor-mTOR (mTORC1) signaling pathway
is a well characterized signaling cascade, which is involved in the
regulation of autophagy (19,20).
The Akt/mTOR signaling pathway in particular has a significant role
in the regulation of autophagy (21,22).
For these reasons, whether PM2.5 regulated autophagy via
activation of Akt/mTOR signaling was evaluated. The phosphorylation
levels of Akt and mTOR were examined by western blotting.
Significant dephosphorylation of Akt and mTOR were detected
following PM2.5 treatment for 12, 24 and 48 h, (Fig. 3A–C; n=3) with the peak effect
occurring following 48 h of exposure. These results indicated that
PM2.5-induced Akt activation may be one of the signaling
pathways, which contribute to the progression of autophagy.
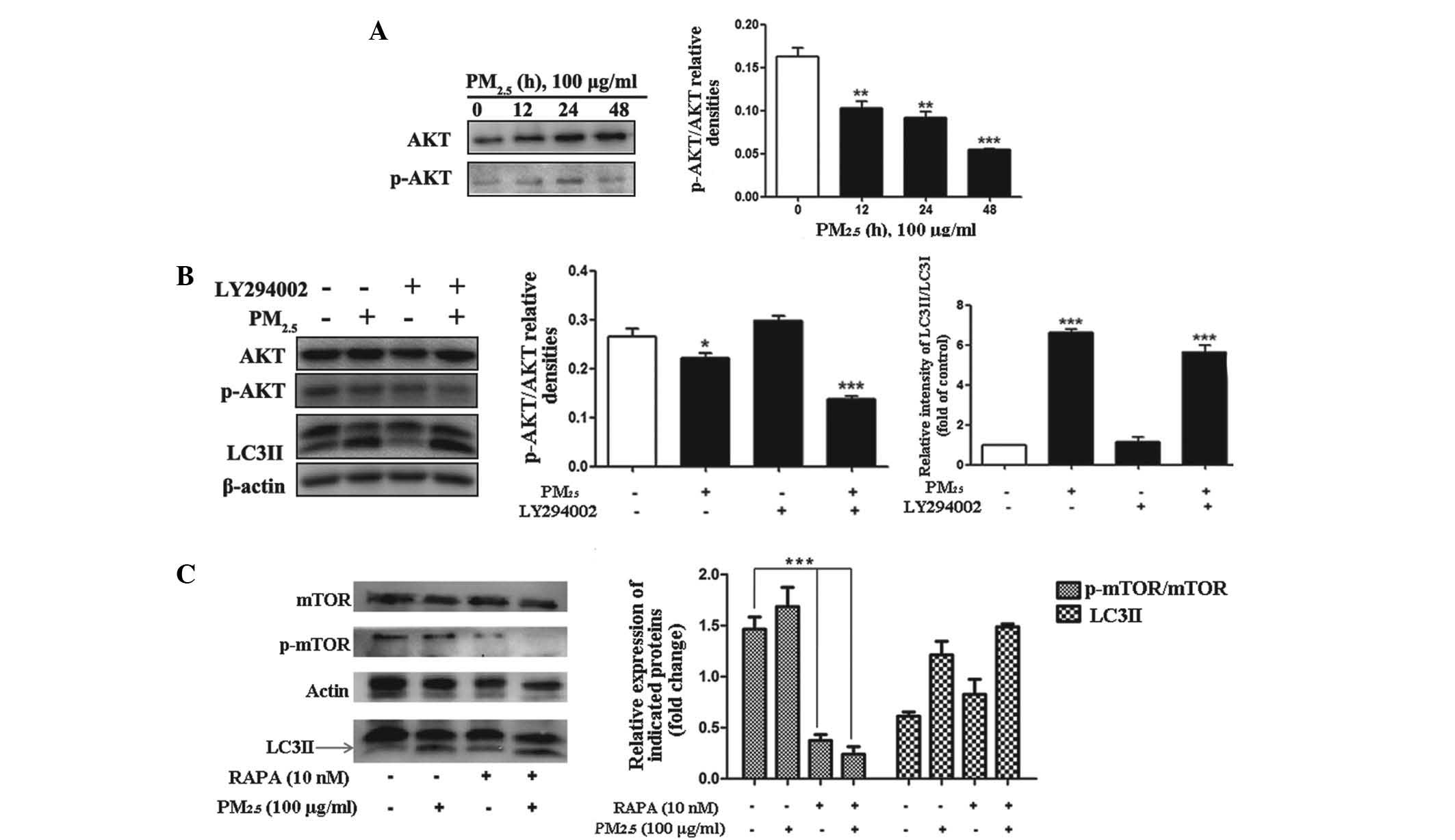 | Figure 3Role of the PI3K/AKT/mTOR signaling
pathways in PM2.5-induced autophagy. Cells were treated
with PM2.5 (0, 12, 24 or 48 h) and the expression levels
of the indicated proteins were analyzed by immunoblotting. (A)
Phosphorylation status of AKT and protein expression levels of AKT
were examined by western blot assay in PM2.5-treated
Beas-2B cells. (B) Cells were treated with 100 µg/ml
PM2.5 for 24 h following 2 h pretreatment with the
inhibitor, LY294002. The phosphorylation status of AKT and protein
expression level of AKT were analyzed by western blotting in
PM2.5-treated Beas-2B cells. (C) Cells were treated with
100 µg/ml PM2.5 for 24 h, following 2 h
pre-treatment with rapamycin. The phosphorylation status of mTOR
and protein expression levels of mTOR were analyzed by western
blotting in PM2.5-treated Beas-2B cells. Values are
expressed as the mean ± standard deviation of three independent
experiments. *P<0.05, **P<0.01 and
***P<0.001 vs. control. PI3K, phosphatidylinositol
3-kinase; mTOR, mammalian target of rapamycin; PM2.5,
particle matter 2.5; p-, phosphorylated; RAPA, rapamycin; LC3II,
microtubule-associated protein 1 light chain 3 II. |
The ERK1/2, but not the JNK or p38 MAPK,
pathway is involved in PM2.5-induced autophagy in
Beas-2B cells
MAPKs, including p38, JNK, and ERK have key roles in
the mediation of autophagy involved in cell death or survival
(23). To explore whether MAPK
signaling pathways were involved in PM2.5-induced
autophagy, the phosphorylation of ERK1/2, JNK and p38 MAPK with LC3
expression and the accumulation of its active form (LC3II) were
evaluated following exposure of Beas-2B cells to 100 µg/ml
PM2.5 for 12, 24 and 48 h. As shown in Fig. 4A, PM2.5 rapidly and
markedly increased ERK phosphorylation following 12 and 24 h of
exposure, though little effect was observed following 48 h of
exposure. To evaluate the involvement of ERK activation in the
modulation of PM2.5-induced autophagy, cells were
pre-treated with ERK inhibitor U0126 and the levels of ERK and
p-ERK were examined by western blot analysis following
PM2.5 exposure. As shown in Fig. 4B, the phosphorylation of ERK was
markedly decreased; however the expression of LC3II was not
significantly altered following U0126 administration with
PM2.5 compared with PM2.5 exposure only.
 | Figure 4Role of MAPK signaling pathways in
PM2.5-induced autophagy (A and C) Western blot assays
were used to examine the total and phosphorylated protein levels of
ERK, JNK and β-actin. (B) Western blot assays were used to examine
the expression of LC3II/LC3I, ERK and p-ERK when ERK inhibitor
U0126 (5 µM) was added for 1 h before PM2.5
treatment. (D) Western blot assays were used to examine the
expression of LC3II/LC3I, p38 and p- p38 when p38 inhibitor
SB203580 (10 µM) was added for 1 h before PM2.5
treatment. (E) Western blot assays were used to examine the
expression of LC3II/LC3I when JNK inhibitor SP600125 (50 µM)
was added for 1 h before PM2.5 treatment. Values are
expressed as the mean ± standard deviation of three independent
experiments. *P<0.05, **P<0.01 and
***P<0.001 vs. control. MAPK, mitogen activated
protein kinase; PM2.5, particle matter 2.5; ERK,
extracellular signal-regulated kinase; JNK, c-Jun N-terminal
kinase; LC3, microtubule-associated protein 1 light chain 3; p-,
phosphorylated. |
JNK and p38-MAPK protein expression levels were also
evaluated by western blotting to determine whether they were
involved in PM2.5-induced autophagy. It was demonstrated
that the phosphorylation of JNK was increased 12 h after
PM2.5 treatment (Fig.
4C). To confirm the underlying mechanism of the JNK and p38
MAPK signaling pathway involvement in PM2.5-induced
autophagy, JNK inhibitor (SP600125) and p38 inhibitor (SB203580)
were applied, following 24 h exposure of Beas-2B cells to 100
µg/ml PM2.5. With regard to p38 (Fig. 4D), pre-treatment with SB203580
decreased the phosphorylation level of p38 and elevated LC3II
expression. As shown in Fig. 4E,
an increase in the conversion of LC3I to LC3II was still observed
after pretreatment with a JNK inhibitor. These data suggested that
with continuous exposure of Beas-2B cells to PM2.5,
p-ERK negatively regulated PM2.5-induced autophagy;
whereas, p-JNK and p38 (of the MAPK signaling pathway) did not have
a significant effect on PM2.5-induced autophagy in
Beas-2B cells.
Expression of p53 in
PM2.5-induced autophagy
To observe whether PM2.5 enhanced the
expression and phosphorylation of p53, immunoblot analysis of
Beas-2B cells following exposure to PM2.5 was performed.
The total protein expression and phosphorylation of p53 were not
significantly altered following PM2.5 exposure (Fig. 5A). To assess the function of p53,
p53 activity was blocked with the p53 inhibitor, PFT-α, in Beas-2B
cells treated for 24 h with or without PM2.5. Western
blot analysis revealed that LC3 protein expression was not markedly
altered by PM2.5 exposure with or without PFT-α
(Fig. 5A). These data indicated
that PM2.5-induced autophagy was p53-independant.
PM2.5-induced Beas-2B cell
autophagy is dependent on reactive oxygen species (ROS)
production
In order to determine whether ROS generation was
involved in the PM2.5-induced autophagy signaling
pathway in Beas-2B cells, LC3II expression was evaluated by western
blot analysis in non-treated cells and in cells pre-incubated with
or without TMTU for 1 h, followed by treatment with 100
µg/ml PM2.5 for 24 h. The activation of LC3II by
PM2.5 was diminished by pre-treatment with TMTU
(Fig. 5B). These data suggested
that PM2.5-induced intracellular autophagy in Beas-2B
cells was dependent on ROS.
Suppression of autophagy enhances
cytoxicity of PM2.5-induced apoptosis in Beas-2B
cells
In order to investigate whether
PM2.5-induced autophagy was involved in apoptosis, PI
staining for apoptosis analysis was performed. Beas-2B cells were
analyzed following treatment with or without autophagy inhibitor
3-MA for 1 h, followed by 100 µg/ml PM2.5
exposure for 24 h. As shown in Fig.
5C, the apoptotic rate of Beas-2B cells in the
PM2.5-treated group was elevated compared with that of
the non-treated group. However, pre-treatment with 3-MA induced an
increase in the level of apoptosis (Fig. 5C). These results suggested that
PM2.5-induced autophagy had a pro-survival function in
Beas-2B cells.
Discussion
Asthma is a chronic inflammatory disease that
influences >300 million individuals worldwide, in which the
majority of cases are characterized by an allergic response,
(24). A previous study provided
evidence demonstrating an association between air pollution and the
development of asthma (25).
Multiple studies, utilizing genetic and histological
approaches, have also indicated that autophagy is associated with
asthma pathogenesis (9,26–28).
The present study aimed to investigate the molecular mechanisms
underlying PM2.5-induced autophagy at a cellular
level.
In the current study, multiple experimental
techniques were used to evaluate PM2.5-induced autophagy
in Beas-2B cells, including fluorescent microscopy, western blot
analysis and TEM, which revealed the formation of characteristic
autophagosomes following 24 h of treatment with PM2.5.
It is well known that the Akt-mTOR signaling pathway is a
significant negative regulator of autophagy, and in the present
study, western blot analyses revealed that PM2.5-induced
autophagy in Beas-2B cells was mediated by dephosphorylation of
Akt/mTOR signaling. Conversely, PM2.5 induced activation
of nutrient sensor AMPK; however, the expression of LC3II was not
reduced upon inhibition of AMPK phosphorylation by DM. These
results suggested that the AMPK pathway was not involved in
PM2.5-mediated induction of autophagy in Beas-2B
cells.
In the present study, PM2.5 was
demonstrated to induce the expression and activation of autophagic
protein LC3II, and to stimulate autophagosome formation, two
characteristics of autophagic pathway initiation. Prolonged
exposure to PM2.5 resulted in time- and dose-dependent
changes in the extent of LC3I to LC3II conversion. In addition,
MAPKs are upstream regulators of mTOR that mediate responses to
various extracellular stimuli (29). All four categories of MAPK (ERK,
p38, JNK/stress-activated protein kinases and big MAPK) have
previously been reported to regulate autophagy (30–32).
Enhanced phosphorylation of the ERK and JNK signaling pathways was
also observed following Beas-2B cell exposure to PM2.5.
Notably, inhibition of ERK activity or expression did not abrogate
PM2.5-induced autophagy in Beas-2B cells. Furthermore,
although the ERK inhibitor effectively blocked the increase in
p-ERK associated with PM2.5 exposure, the expression of
LC3II remained unchanged by this inhibition, indicating that the
MEK-ERK1/2 pathway was not involved in autophagy induction by
PM2.5.
Whether the JNK and p38 MAPK signaling pathways were
involved in PM2.5-induced autophagy in Beas-2B cells was
also examined. The phosphorylation of JNK and p38 MAPK was
evaluated, but neither was found to be significantly involved in
PM2.5-induced autophagy in Beas-2B cells, indicating a
lack of MAPK signaling significance in this process.
In the present study, it was revealed that Akt/mTOR
had a negatively regulatory role in PM2.5-induced
autophagy in Beas-2B cells. However, this autophagy was independent
of JNK, concurrent with previous findings (33–35),
indicating that a different MAPK response to PM2.5 is
dependent on cell-type specification.
Urich et al (36) examined PM2.5-induced
apoptosis in the alveolar epithelium, and identified that it
required transcriptional activation of p53 and its phosphorylation;
however, the results of the present study did not indicate that p53
was activated by PM2.5 in Beas-2B cells. The autophagy
induced by PM2.5 exposure was also demonstrated to be
independent of p53. p53 has previously been shown to serve a dual
role in the control of autophagy (37,38).
In addition, it was demonstrated that ROS scavenger TMTU was able
to diminish the expression of LC3II protein induced by
PM2.5, indicating that ROS were involved in
PM2.5-induced autophagy. Previous studies have
demonstrated that apoptosis is implicated in asthma pathogenesis
(39,40) and, in the context of
PM2.5 exposure, it was demonstrated that pre-treatment
of Beas-2B cells with 3-MA enhanced PM2.5-inducible
apoptosis. These results indicated that the autophagy induced by
PM2.5 had a pro-survival function in Beas-2B cells.
Together, the results of the present study indicated that multiple
autophagy-associated signaling pathways are activated following
PM2.5 exposure, and that alterations in the Akt-mTOR
inhibition pathway likely have major roles in the induction of
autophagy.
The role of autophagy in the regulation of Beas-2B
cell survival and apoptosis is complex. Further studies are
required in order to characterize the specific autophagic signaling
pathways activated by exposure to PM2.5 (for example,
macroautophagy, microautophagy and chaperone-mediated autophagy) in
isolated primary mouse bronchial epithelial cells. Although the
evaluation of PM2.5-induced autophagy in Beas-2B cells
in vitro is not necessarily indicative of the conditions
in vivo, particularly in the context of human diseases, it
was hypothesized that PM2.5-induced autophagy may be a
pro-survival response to PM2.5 exposure. Additional
studies are required in order to elucidate the molecular mechanisms
underlying PM2.5-induced autophagy and apoptosis in the
context of health and disease.
In conclusion, the results of the present study
indicated that PM2.5 exposure stimulated autophagy in
human bronchial epithelial cells, identified by ultrastructural and
biochemical features of autophagy. PM2.5-induced
autophagy was also demonstrated to be associated with altered
signaling via the Akt/mTOR pathway, but was independent of p53.
Furthermore, PM2.5-induced autophagy was regulated in
part by ROS-associated mechanisms. However, the specific molecular
mechanisms underlying the in vivo significance of these
results remain to be elucidated. The results of the present study
provide a mechanistic basis for the development of clinical
applications targeting these signaling pathways for the prevention
and/or treatment of PM2.5-induced lung disease.
Acknowledgments
The present study was supported by the Science and
Technology Innovation Fund of Guangdong Medical College (no.
STIF201109), the Natural Science Foundation of China project (no.
NSFC81172615), Guangdong Natural Science Foundation (no.
S20122010008299) and the Science and Technology Planning Project of
Guangdong Province (no. 2012B031800223).
Abbreviations:
|
mTOR
|
mammalian target of rapamycin
|
|
LC3
|
microtubule-associated protein 1 light
chain 3
|
|
AMPK
|
adenosine monophosphate-activated
protein kinase
|
|
p38
|
p38 mitogen-activated protein
kinase
|
|
JNK
|
c-Jun N-terminal kinase
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
MAPK
|
mitogen-activated protein kinases
|
|
3-MA
|
3-methyladenine
|
|
PM2.5
|
particulate matter 2.5
|
References
|
1
|
Zanobetti A, Franklin M, Koutrakis P and
Schwartz J: Fine particulate air pollution and its components in
association with cause-specific emergency admissions. Environ
Health. 8:582009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Delfino RJ, Sioutas C and Malik S:
Potential role of ultrafine particles in associations between
airborne particle mass and cardiovascular health. Environ Health
Perspect. 113:934–946. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dockery DW, Pope CA III, Xu X, et al: An
association between air pollution and mortality in six U.S. cities.
N Engl J Med. 329:1753–1759. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Trejo Bittar HE, Yousem SA and Wenzel SE:
Pathobiology of severe asthma. Annu Rev Pathol. 10:511–545. 2015.
View Article : Google Scholar
|
|
5
|
Mehta M, Chen LC, Gordon T, Rom W and Tang
MS: Particulate matter inhibits DNA repair and enhances
mutagenesis. Mutat Res. 657:116–121. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nordling E, Berglind N, Melén E, et al:
Traffic-related air pollution and childhood respiratory symptoms,
function and allergies. Epidemiology. 19:401–408. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Anderson HR, Favarato G and Atkinson RW:
Long-term exposure to air pollution and the incidence of asthma:
meta-analysis of cohort studies. Air Qual Atmos Health. 6:47–56.
2013. View Article : Google Scholar
|
|
8
|
Zeng Y, Yang X, Wang J, Fan J, Kong Q and
Yu X: Aristolochic acid I induced autophagy extenuates cell
apoptosis via ERK 1/2 pathway in renal tubular epithelial cells.
PloS One. 7:e303122012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Poon AH, Chouiali F, Tse SM, et al:
Genetic and histologic evidence for autophagy in asthma
pathogenesis. J Allergy Clin Immunol. 129:569–571. 2012. View Article : Google Scholar :
|
|
10
|
Maiuri MC, Galluzzi L, Morselli E, Kepp O,
Malik SA and Kroemer G: Autophagy regulation by p53. Curr Opin Cell
Biol. 22:181–185. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Glick D, Barth S and Macleod KF:
Autophagy: cellular and molecular mechanisms. J Pathol. 221:3–12.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Akhtar S, Yousif MH, Chandrasekhar B and
Benter IF: Activation of EGFR/ERBB2 via pathways involving ERK1/2,
P38 MAPK, AKT and FOXO enhances recovery of diabetic hearts from
ischemia-reperfusion injury. PloS One. 7:e390662012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Soberanes S, Panduri V, Mutlu GM, Ghio A,
Bundinger GR and Kamp DW: p53 mediates particulate matter-induced
alveolar epithelial cell mitochondria-regulated apoptosis. Am J
Respir Crit Care Med. 174:1229–1238. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lin Z, Liu T, et al: AKT/mTOR and c-Jun
N-terminal kinase signaling pathways are required for chrysotile
asbestos-induced autophagy. Free Radic Biol Med. 72:296–307. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang L, Liu G, Lin Z, et al:
Pro-inflammatory response and oxidative stress induced by specific
components in ambient particulate matter in human bronchial
epithelial cells. Environ Toxicol. Dec 23–2014.Epub ahead of print.
View Article : Google Scholar
|
|
16
|
Wiltfang J, Smirnov A, Schnierstein B, et
al: Improved electrophoretic separation and immunoblotting of
beta-amyloid (A beta) peptides 1–40, 1–42, and 1–43.
Electrophoresis. 18:527–532. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhou C, Zhong W, Zhou J, et al: Monitoring
autophagic flux by an improved tandem fluorescent-tagged LC3
(mTagRFP-mWasabi-LC3) reveals that high-dose rapamycin impairs
autophagic flux in cancer cells. Autophagy. 8:1215–1226. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bolster DR, Crozier SJ, Kimball SR and
Jefferson LS: AMP-activated protein kinase suppresses protein
synthesis in rat skeletal muscle through down-regulated mammalian
target of rapamycin (mTOR) signaling. J Biol Chem. 277:23977–23980.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Corcelle EA, Puustinen P and Jäättelä M:
Apoptosis and autophagy: Targeting autophagy signalling in cancer
cells – ‘trick or treats’? FEBS J. 276:6084–6096. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jung CH, Ro SH, Cao J, Otto NM and Kim DH:
mTOR regulation of autophagy. FEBS Lett. 584:1287–1295. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Codogno P and Meijer AJ: Autophagy and
signaling: their role in cell survival and cell death. Cell Death
Differ. 12(Suppl 2): 1509–1518. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Roy B, Pattanaik AK, Das J, et al: Role of
PI3K/Akt/mTOR and MEK/ERK pathway in Concanavalin A induced
autophagy in HeLa cells. Chem Biol Interact. 210:96–102. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hamamura K, Goldring MB and Yokota H:
Involvement of p38 MAPK in regulation of MMP13 mRNA in chondrocytes
in response to surviving stress to endoplasmic reticulum. Arch Oral
Biol. 54:279–286. 2009. View Article : Google Scholar :
|
|
24
|
Dougherty R and Fahy JV: Acute
exacerbations of asthma: epidemiology, biology and the
exacerbation-prone phenotype. Clin Exp Allergy. 39:193–202. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Delfino RJ, Wu J, Tjoa T, Gullesserian SK,
Nickerson B and Gillen DL: Asthma morbidity and ambient air
pollution: effect modification by residential traffic-related air
pollution. Epidemiology. 25:48–57. 2014. View Article : Google Scholar
|
|
26
|
Jyothula SS and Eissa NT: Autophagy and
role in asthma. Curr Opin Pulm Med. 19:30–35. 2013. View Article : Google Scholar
|
|
27
|
Martin LJ, Gupta J, Jyothula SS, et al:
Functional variant in the autophagy-related 5 gene promotor is
associated with childhood asthma. PLoS One. 7:e334542012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Poon A, Eidelman D, Laprise C and Hamid Q:
ATG5, autophagy and lung function in asthma. Autophagy. 8:694–695.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Albert L, Karsy M, Murali R and
Jhanwar-Uniyal M: Inhibition of mTOR activates the MAPK pathway in
glioblastoma multiforme. Cancer Genomics Proteomics. 6:255–261.
2009.PubMed/NCBI
|
|
30
|
Hazzalin CA and Mahadevan LC:
MAPK-regulated transcription: a continuously variable gene switch?
Nat Rev Mol Cell Biol. 3:30–40. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Webber JL and Tooze SA: Coordinated
regulation of autophagy by p38alpha MAPK through mAtg9 and p38IP.
EMBO J. 29:27–40. 2010. View Article : Google Scholar :
|
|
32
|
de la Cruz-Morcillo MA, Valero ML,
Callejas-Valera JL, et al: P38MAPK is a major determinant of the
balance between apoptosis and autophagy triggered by
5-fluorouracil: implication in resistance. Oncogene. 31:1073–1085.
2012. View Article : Google Scholar
|
|
33
|
Brigelius-Flohé R: Commentary: oxidative
stress reconsidered. Genes Nutr. 4:161–163. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cheng Y, Qiu F, Tashiro S, Onodera S and
Ikejima T: ERK and JNK mediate TNFalpha-induced p53 activation in
apoptotic and autophagic L929 cell death. Biochem Biophys Res
Commun. 376:483–488. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kabesch M and Adcock IM: Epigenetics in
asthma and COPD. Biochimie. 94:2231–2241. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Urich D, Soberanes S, Burgess Z, et al:
Proapoptotic Noxa is required for particulate matter-induced cell
death and lung inflammation. FASEB J. 23:2055–2064. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
McCarthy N: Autophagy: Directed
development. Nat Rev Cancer. 14:74–75. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Green DR and Kroemer G: Cytoplasmic
functions of the tumour suppressor p53. Nature. 458:1127–1130.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kim MN, Lee KE, Hong JY, et al:
Involvement of the MAPK and PI3K pathways in chitinase 3-like
1-regulated hyperoxia-induced airway epithelial cell death. Biochem
Biophys Res Commun. 421:790–796. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Takada E, Furuhata M, Nakae S, Ichijo H,
Sudo K and Mizuguchi J: Requirement of apoptosis-inducing kinase 1
for the induction of bronchial asthma following stimulation with
ovalbumin. Int Arch Allergy Immunol. 162:104–114. 2013. View Article : Google Scholar : PubMed/NCBI
|
















