Introduction
Gliomas are the most common type of primary tumor of
the brain. They are classified into four clinical grades, according
to histology and prognosis, by the World Health Organization
(1). The most malignant form,
Grade IV glioma, is glioblastoma multiforme (GBM). The survival
rate of patients with GBM, has not improved since the 1980s, with a
1-year relative survival rate of ~30% and a 5-year survival rate of
<5% (2). Even with
comprehensive treatment, which includes surgery, chemotherapy and
irradiation, the prognosis and treatment of GBMs remains limited
(3).
Among several inflammatory cytokines, the
transforming growth factor (TGF)β family has been implicated in
glioma. There are five subclasses of the TGFβ family: β1, β2, β3,
β4 and β5. TGFβ1, β2 and β3 are expressed in mammalian tissues
(4). Within the TGFβ family, TGFβ2
is the most potent factor, which is involved in the initiation and
maintenance of GBM (5).
TGFβ family cytokines affect a diverse array of
cellular processes, including cellular proliferation,
differentiation, apoptosis and migration (6). Once activated, TGFβ binds and
activates the TGFβ type II receptor, TβRII, and the TGFβ type I
receptor, TβRI, which phosphorylates Smad2 and Smad3 to generate
phosphorylated (p-)Smad2 and p-Smad3. Upon phosphorylation, Smad2
and Smad3 form transcriptional complexes with Smad4 and other
transcription factors and accumulate in the nucleus, where they
regulate transcription (7,8).
The Smad family of proteins can be divided into
three different subfamilies: The receptor-activated Smads
(R-Smads), common mediator Smads and inhibitory Smads (9). Smad2 and Smad3 are R-Smads and are
directly phosphorylated in response to TGFβ and activin (10). Although Smad2 and Smad3 belong to
the R-Smad subfamily and are important in the biological effects of
TGFβ, they do not have identical effects in cellular
proliferation.
A previous study, involving a human lens cell line,
revealed that the effect of TGFβ2 on cell proliferation depends on
the Smad3 signaling pathway (11).
Another study found that Smad2 and Smad3 have different roles in
pancreatic ductal adenocarcinoma cells (12).
As mentioned above, Smad2 and Smad3 have different
functions, however, the specific functions of Smad2 and Smad3 in
GBM remain to be elucidated, as does whether each response is
mediated predominantly or exclusively by only one of the two
R-Smads. The predominant focus of the present study was to
determine the specific roles of Smad2 and Smad3 in the
proliferative effect of TGFβ in GBM.
Therefore, the present study investigated how
downregulation in the expression levels of Smad2 and Smad3 affected
glioma cell proliferation, and whether GBM cell proliferation was
controlled differentially by Smad2 and Smad3. For this purpose, the
well-characterized U251 GBM cell line, which has retained a
functional TGFβ2/Smad pathway, was used (13). RNA interference was used to
specifically knock down the expression of the two R-Smads to
determine whether TGFβ2 proliferation was dependent on the Smad3
and/or Smad2 signaling pathway.
Materials and methods
Cell lines and cell culture
U251 cells were purchased from American Type Culture
Collection (Manassas, VA, USA). The U251 cells were cultured in
Dulbecco’s modified Eagle’s medium (DMEM; Gibco-BRL, Grand Island,
NY, USA), supplemented with 10% fetal bovine serum (FBS; Gibco-BRL)
at 37°C in a humidified 5% CO2 atmosphere.
Short hairpin (sh)RNA transfection
The cells were grown to 70–90% confluence, at the
time of transfection. Smad2 and Smad3 shRNAs were synthesized and
inserted into the pGPU6/GFP/Neo empty vector plasmid to construct
the pGPU6/GFP/Neo-Smad2-shRNA (psh-Smad2) and
pGPU6/GFP/Neo-Smad3-shRNA (psh-Smad3) plasmids by Shanghai
GenePharma (Shanghai, China). The pGPU6/GFP/Neo-negative
control-shRNA (psh-NC) non-silencing control plasmid was also
synthesized by GenePharma and was confirmed by BLAST analysis
(http://blast.ncbi.nlm.nih.gov.ezp.lib.unimelb.edu.au/Blast.cgi)
to have no complementarity to any mammalian mRNA sequence. The
following gene-specific sequences were used:
5′-GCAGAACTATCTCCTACTACT-3′ for Smad2, 5′-GGCTGCTCTCCAATGTCAACA-3′
for Smad3 and 5′-GTTCTCCGAACGTGTCACGT-3′ for NC shRNA. The
gene-specific sequences were generated by Shanghai GenePharma. The
plasmids were transfected using Lipofectamine® 2000
(Invitrogen Life Technologies, Carlsbad, CA, USA), according to the
manufacturer’s instructions. Transfection efficiency was determined
using an inverted microscope (DMI3000B; Leica Microsystems,
Wetzlar, Germany).
Total RNA extraction, cDNA synthesis and
reverse transcription-quantitative polymerase chain reaction
(RT-qPCR)
The expression levels of Smad2 and Smad3 were
determined by qPCR. The cells were collected 24 h post-transfection
(80–90% confluence). The total RNA from the cultured tumor cells
was isolated using TRIzol reagent, according to the manufacturer’s
protocol (Invitrogen Life Technologies). RT was performed to
generate cDNA using the PrimeScript® RT-PCR kit (Takara
Bio, Inc., Otsu, Japan).
For qPCR, SYBR® Premix Ex Taq™ II (Takara
Bio, Inc.) was used, according to the manufacturer’s instructions,
using a 7500 Fast Real-Time PCR system (Applied Biosystems, Foster
City, CA, USA). Each 20 μl reaction contained ~100 ng DNA
template and 0.4 μM of the forward and reverse primers
(Invitrogen Life Technologies). The GAPDH gene served as an
endogenous reference. The primer sequences used in the present
study are listed in Table I.
 | Table IPrimers used for reverse
trasnscription-quantitative polymerase chain reaction. |
Table I
Primers used for reverse
trasnscription-quantitative polymerase chain reaction.
| Primer | Sequence | Product length
(bp) |
|---|
| Smad2 | F:
5′-ACTAACTTCCCAGCAGGAAT-3′
R: 5′-GTTGGTCACTTGTTTCTCCA-3′ | 40 |
| Smad3 | F:
5′-CCACGCAGAACGTCAACA-3′
R: 5′-TTGAAGGCGAACTCACACAG-3′ | 38 |
| GAPDH | F:
5′-GAGTCAACGGATTTGGTCGT-3′
R: 5′-TTGATTTTGGAGGGATCTCG-3′ | 40 |
The thermocycler parameters were as follows: 95°C
for 30 sec, followed by 40 cycles of 95°C for 3 sec and 56°C for 30
sec. The qPCR reactions were performed in triplicate and the
relative mRNA expression levels were quantified based on the
threshold cycle (Ct) value, normalized to GAPDH, and expressed as
relative quantities. The quantity of the target normalized to GAPDH
and relative to the calibrator target gene in the TGFβ2 group, was
calculated using the following formula: Fold-change in TGFβ2 (+/−)
= 2−ΔΔCT, where ΔΔCT = ΔCTT −
ΔCTC (ΔCT = CTtarget −
CTreference) (14).
Where ΔCTT represents the ΔCt value of the target, and
ΔCTC represents the ΔCt value of the control.
Western blot analysis
At 24 h post-transfection, protein was extracted
from the cultured cells using radioimmunoprecipitation assay lysis
buffer containing 1% protease inhibitors (Roche Diagnostics, Basel,
Switzerland), the cellular proteins were then boiled at 100°C for
10 min. The protein concentration was determined using Thermo
Scientific NanoDrop 2000 (Thermo Scientific, Waltham, MA, USA).
Equal quantities of protein (50 ng) were subjected to a 10%
SDS-PAGE (Applygen Technologies, Inc., Beijing, China) and then
transferred to a nitrocellulose membrane (Millipore, Bedford, MA,
USA). The membranes were blocked with 5% skim milk (Applygen
Technologies, Inc.) for 2 h at room temperature and incubated with
rabbit anti-human monoclonal Smad2 (cat. no. 3122) and Smad3 (cat.
no. 9523) antibodies (1:1,000; Cell Signalling Technology, Inc.,
Heidelberg, Germany) or GAPDH antibody (1:1,000; cat. no. 2118;
Cell Signalling Technology, Inc.) in 5% skim milk overnight at 4°C.
Following incubation, the membranes were washed three times in
phosphate-buffered saline (PBS) containing 0.1% Tween-20 (PBST) for
10 min. The membranes were then incubated with a goat anti-rabbit
antibody (1:1,000; cat. no. ZDR-5306; Applygen Technologies, Inc.)
for 90 min at room temperature. Following three further washes in
PBST, the protein expression levels were determined by enhanced
chemiluminescence (Applygen Technologies, Inc.) and exposure to
chemiluminescent film (Applygen Technologies, Inc.).
Cell proliferation assay
At 24 h post-transfection, a Cell Counting kit-8
(CCK-8; Dojindo Molecular Technologies, Kunamoto, Japan) was used
to determine the effect of TGFβ2 (R&D Systems, Inc., Wiesbaden,
Germany) on the proliferation of the transfected cells. The cells
(3×103) cells were seeded in 96-well plates and cultured
in DMEM without FBS at 37°C for 24 h. The culture medium was then
removed. Subsequently, the cells were treated with or without 1.3
ng/ml TGFβ2 in DMEM for 12 h at 37°C. Subsequently, 10 μl
CCK-8 dye was add to each well and the cells were incubated at 37°C
for 1 h. The absorbance at 450 nm was determined using a multimode
reader (Wellscan MK3; Labsystems Dragon, Helsinki, Finland). Three
parallel experiments for each sample were used to assess cell
proliferation.
Statistical analysis
Statistical analyses were performed using the SPSS
software package (version 16.0; SPSS, Inc., Chicago, IL, USA). The
results were analyzed using one-way analysis of variance and a
least significant difference t-test at a global level of
significance of 95%. The data are presented as the mean ± standard
deviation. P<0.05 was considered to indicate a statistically
significant difference.
Results
Specific silencing of the TGFβ2/Smad2 or
TGFβ2/Smad3 signaling pathway using shRNA
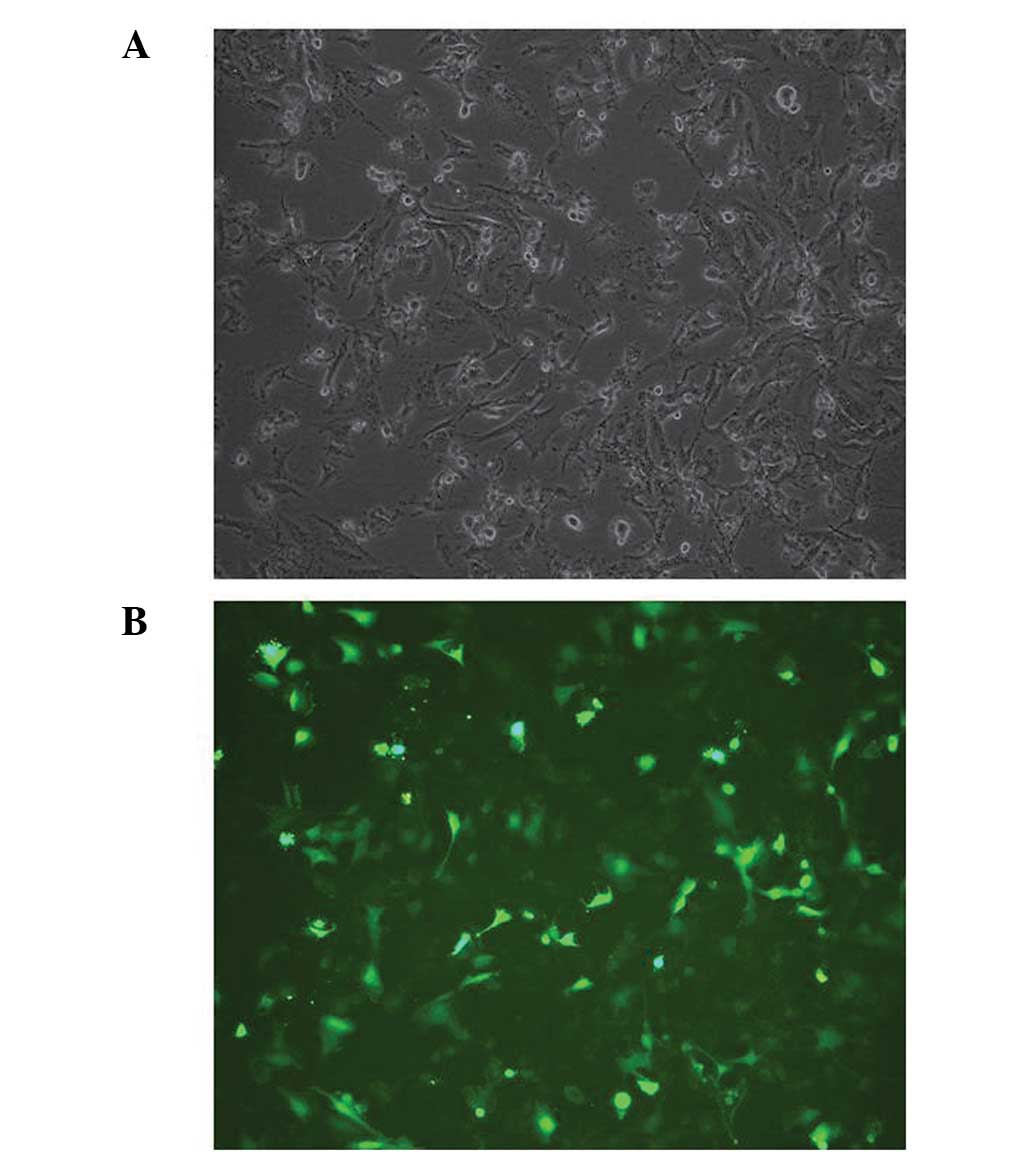
To confirm the effect of the constructed shRNAs, the
shRNA expression vectors were transfected into U251 cells. The
transfection efficiency was confirmed using an inverted microscope,
and the results demonstrated that the pGPU6/GFP/Neo vector, which
contained a cassette of GFP, had been successfully transfected into
the U251 cells (Fig. 1). The
expression levels of Smad2 and Smad3 were detected using an RT-qPCR
assay and western blot analysis. The psh-NC plasmid was used as a
control. The results of the qPCR and western blot analyses revealed
that Smad2 and Smad3 were specifically and efficiently knocked down
by their corresponding shRNA (Fig.
2).
Depletion of Smad2/3 enhances cell
proliferation
The present study investigated how downregulation of
the expression levels of Smad2 and Smad3 affected glioma cell
proliferation. Therefore, following transfection of the cells with
Smad2 shRNA or Smad3 shRNA, the cell growth response to 24 h
treatment with DMEM was determined using a CCK-8 assay. A
significant increase in absorbency at 450 nm was observed in the
U251 cells transfected with Smad2 shRNA (P<0.000) or Smad3 shRNA
(P<0.000; Fig. 3). This result
suggested that silencing Smad2 or Smad3 may promote cell
proliferation.
Induction of cell proliferation by TGFβ2
depends on the TGFβ2/Smad3 signaling pathway
To determine the optimal concentration of TGFβ2 in
the present study, the U251 cells were treated with increasing
doses of TGFβ2 for 24 h and rapid TGFβ2 responses, which were more
likely to be direct, were determined to identify the optimal
concentration of 1.3 ng/ml (Fig.
4A).
It is widely accepted that the TGFβ2/Smads signaling
pathway is involved in the regulation of cell proliferation
(15). However, the relative
involvement of Smad2 and Smad3 in the control of TGFβ2-induced cell
proliferation in glioma cells remains to be elucidated. Therefore,
the present study investigated the mechanism underlying the
induction of proliferation by TGFβ2/Smads in glioma. The U251 cells
were transfected with Smad2 shRNA, Smad3 shRNA or NC shRNA, and the
growth response following 12 h treatment with or without TGFβ2 was
assessed using a CCK-8 assay. The proliferation rates of the cells
transfected with Smad2 shRNA or NC shRNA increased in the presence
of TGFβ2 (P=0.001 and P=0.009, respectively). However, the rate of
cell proliferation was similar between the cells treated with or
without TGFβ2 when the Smad3 signaling pathway was inhibited
(P=0.258; Fig. 4B). These results
demonstrated that the promoting effect of TGFβ2 on cell
proliferation was dependent on the Smad3 signaling pathway.
Discussion
Glioma is the most common type of primary
intracranial malignancy, and GBM is the most malignant type of
gliom, accounting for ~70% of malignant brain tumors in adults
(16). Despite advances in
treatment, including surgical resection followed by concurrent
chemotherapy with radiation, GBM remains an incurable and
life-threatening disease, with a median survival rate of ~9–15
months following diagnosis (17).
TGFβ proteins regulate cell function and are important in
development and carcinogenesis (18). Although the downstream signaling
events, which occur to stimulate cell proliferation remain to be
fully elucidated, the intracellular effectors of TGFβ signaling,
the Smad proteins, are activated by receptors and are translocated
into the nucleus, where they regulate transcription (19).
Previous studies have investigated the expression
levels of Smad2 and Smad3 in gliomas in tumor specimens and cell
lines, however, the results have been inconsistent. Zhang et
al examined the expression levels of downstream components of
the TGFβ receptor, including Smad2 and Smad3, in 10 glioma cell
lines. The results revealed that the protein expression levels of
Smad2 and Smad3 were lower in the glioma cell lines compared with
normal astrocytes (20).
Similarly, Kjellman et al analyzed the mRNA expression
levels of Smad2 and Smad3 in tissue specimens from 23 cases of
glioma, in which decreased mRNA levels of Smad2 and Smad3 were
observed and correlated with the degree of malignancy (5). However, a study by Horst H et
al found that the mRNA expression levels of Smad2 and Smad3
increased with the degree of malignancy (21).
To examine the effects of the downstream components
of the TGFβ2/Smads signaling pathway, Smad2 and Smad3, on GBM cell
proliferation, the present study transfected U251 cells with
shRNAs, to selectively deplete Smad2 and Smad3, and measured the
growth of the cells. The results revealed that the knock down of
Smad2 and Smad3 enhanced cellular proliferation, demonstrating that
Smad2 and Smad3 had an inhibitory effect on cell proliferation in
this glioma cell line. A study by Zhang et al demonstrated
that the protein expression levels of Smad2 and Smad3 were lower in
glioma cell lines compared with normal astrocytes (20). Considering these results, the
preset study hypothesized that the ability to resist TGFβ2-mediated
growth inhibition in malignant glioma cells was due to a decrease
in the expression levels of Smad2 and Smad3 in the TGFβ2 signaling
pathway.
There is now substantial evidence that Smad2 and
Smad3 have distinct functions in TGFβ signaling. Inhibiting the
function of endogenous Smad3 in ductal adenocarcinoma, liver and
human lens cell lines significantly suppresses the effect of TGFβ
on cell proliferation (11,12,20).
However, there is no evidence that Smad2 and Smad3 have distinct
functions in GBM growth.
The present study aimed to confirm the functions of
Smad2 and Smad3 in GBM cells by transfecting U251 cells with shRNAs
to selectively deplete Smad2 and Smad3, and analyzing the
proliferative response of the cells to TGFβ2 using a CCK-8 assay.
The results revealed a difference in the rate of cell proliferation
between the cells with and without TGFβ2 following transfection
with Smad2 shRNA or NC shRNA. However, the rate of cell
proliferation was similar between the cells treated with and
without TGFβ2 when the Smad3 signaling pathways were inhibited.
These results demonstrated that Smad3 was more important in the
regulation of TGFβ2-inducedf cell proliferation in glioma cells.
Previous studies have reported that Smad2 contains an extra exon in
the MH1 domain, absent from Smad3, which encodes 30 amino acids and
interferes with DNA recognition; thus, Smad3 can interact directly
with Smad-binding element sequences in DNA (22). Based on these studies and the
results of the present study, we hypothesized that the differences
in the molecular structures of Smad2 and Smad3 may be the reason
underlying why Smad3 has a significant effect on the regulation of
TGFβ2-induced cell proliferation in glioma cells. However, further
investigations experiments are required to confirm these
findings.
In conclusion, the present study provided evidence
that the Smad3 pathway is important in malignant glioma cells and
suggested that Smad2 and Smad3 have tumor suppressor activities.
Therefore, the proliferation of GBM cannot be prevented by
inhibiting the TGFβ2/Smad2 and 3 signaling pathway. Although
further studies are required, these results may provide a reference
in attempts to modulate the growth of malignant gliomas.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (nos. 81071776 and 81372354), the
Beijing Natural Science Foundation (nos. 7102027, and 7132034) and
the Beijing Health System High-level Personnel Building Foundation
(no. 2013-2-018).
References
|
1
|
Brat DJ, Scheithauer BW, Fuller GN and
Tihan T: Newly codified glial neoplasms of the 2007 WHO
Classification of Tumours of the Central Nervous System:
angiocentric glioma, pilomyxoid astrocytoma and pituicytoma. Brain
Pathol. 17:319–324. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Deorah S, Lynch CF, Sibenaller ZA and
Ryken TC: Trends in brain cancer incidence and survival in the
United States: Surveillance, Epidemiology, and End Results Program,
1973 to 2001. Neurosurg Focus. 20:E12006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Eisele G and Weller M: Targeting apoptosis
pathways in glioblastoma. Cancer Lett. 332:335–345. 2013.
View Article : Google Scholar
|
|
4
|
Tanihara H, Inatani M and Honda Y: Growth
factors and their receptors in the retina and pigment epithelium.
Prog Retin Eye Res. 16:271–301. 1997. View Article : Google Scholar
|
|
5
|
Kjellman C, Olofsson SP, Hansson O, et al:
Expression of TGF-beta isoforms, TGF-beta receptors, and Smad
molecules at different stages of human glioma. Int J Cancer.
89:251–258. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Joseph JV, Balasubramaniyan V, Walenkamp A
and Kruyt FA: TGF-beta as a therapeutic target in high grade
gliomas- Promises and challenges. Biochem Pharmacol. 85:478–485.
2013. View Article : Google Scholar
|
|
7
|
Massagué J, Seoane J and Wotton D: Smad
transcription factors. Genes Dev. 19:2783–2810. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Inman GJ and Hill CS: Stoichiometry of
active smad-transcription factor complexes on DNA. J Biol Chem.
277:51008–51016. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mishra L, Shetty K, Tang Y, Stuart A and
Byers SW: The role of TGF-beta and Wnt signaling in
gastrointestinal stem cells and cancer. Oncogene. 24:5775–5789.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shi Y and Massagué J: Mechanisms of
TGF-beta signaling from cell membrane to the nucleus. Cell.
113:685–700. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li J, Tang X and Chen X: Comparative
effects of TGF-beta2/Smad2 and TGF-beta2/Smad3 signaling pathways
on proliferation, migration, and extracellular matrix production in
a human lens cell line. Exp Eye Res. 92:173–179. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ungefroren H, Groth S, Sebens S, Lehnert
H, Gieseler F and Fändrich F: Differential roles of Smad2 and Smad3
in the regulation of TGF-beta1-mediated growth inhibition and cell
migration in pancreatic ductal adenocarcinoma cells: control by
Rac1. Mol Cancer. 10:672011. View Article : Google Scholar
|
|
13
|
Bruna A, Darken RS, Rojo F, et al: High
TGFbeta-Smad activity confers poor prognosis in glioma patients and
promotes cell proliferation depending on the methylation of the
PDGF-B gene. Cancer Cell. 11:147–160. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zheng W, Zhao X, Wang J and Lu L: Retinal
vascular leakage occurring in GABA Rho-1 subunit deficient mice.
Exp Eye Res. 90:634–640. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Feng C and Zuo Z: Regulatory factor
X1-induced down-regulation of transforming growth factor beta2
transcription in human neuroblastoma cells. J Biol Chem.
287:22730–22739. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schwartzbaum JA, Fisher JL, Aldape KD and
Wrensch M: Epidemiology and molecular pathology of glioma. Nat Clin
Pract Neurol. 2:494–503. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Stupp R, Mason WP, van den Bent MJ, et al:
Radiotherapy plus concomitant and adjuvant temozolomide for
glioblastoma. Engl J Med. 352:987–996. 2005. View Article : Google Scholar
|
|
18
|
Tirado-Rodriguez B, Ortega E,
Segura-Medina P and Huerta-Yepez S: TGF-β: An important mediator of
allergic disease and a molecule with dual activity in cancer
development. J Immunol Res. 2014:3184812014. View Article : Google Scholar
|
|
19
|
Der ynck R: Smad-dependent and
Smad-independent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar
|
|
20
|
Zhang L, Sato E, Amagasaki K, Nakao A and
Naganuma H: Participation of an abnormality in the transforming
growth factor-beta signaling pathway in resistance of malignant
glioma cells to growth inhibition induced by that factor. J
Neurosurg. 105:119–128. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Horst HA, Scheithauer BW, Kelly PJ and
Kovach JS: Distribution of transforming growth factor-beta(1) in
human astrocytomas. Hum Pathol. 23:1284–1288. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dennler S, Huet S and Gauthier JM: A short
amino-acid sequence in MH1 domain is responsible for functional
differences between Smad2 and Smad3. Oncogene. 18:1643–1648. 1999.
View Article : Google Scholar : PubMed/NCBI
|