Introduction
Migraine is a complex neurovascular disorder that is
often manifested as severe, episodic and predominantly unilateral
throbbing head pain with hypersensitivity to light, sound and
movement (1–3), affecting ~15% of the adult population
worldwide, and can lead to ischemic stroke, depression, cognitive
impairment and epilepsy (3).
However, the mechanisms underlying migraine are not completely
understood. The trigeminovascular system (TVS) mediates neurogenic
inflammation, which is characterized by meningeal vascular
expansion, plasma protein leakage and mast cell degranulation
(4).
Another important symptom, which is often observed
in patients suffering from chronic migraine, is cutaneous allodynia
(5). Cutaneous allodynia is
believed to be a result of central sensitization, which is also
mediated by the TVS (6,7). The neurons in the spinal trigeminal
nucleus caudalis (TNC) receive input signals from the dura mater
and periorbital skin (2). As a
result of this sensitization, non-noxious stimuli of the skin are
perceived as painful (5). However,
limited information is available on the tissue factors that
participate in central sensitization and the mechanisms that
maintain the activation of meningeal nociceptors that cause
neurogenic inflammation and sensitization.
Extracellular signal-regulated protein kinases (ERK)
are mitogen-activated protein kinases that are activated by
membrane depolarization and calcium influx (8), and known to be one of the
intracellular signaling pathways involved in neuronal plasticity
(9,10). The phosphorylation of ERK (p-ERK)
is a response to the noxious stimulation of peripheral transient
receptor potential vanilloid receptor 1 (TRPV1) (11). The noxious information is carried
to the peripheral nerve endings and the TNC.
Calcitonin gene-related peptide (CGRP) is a key
neuropeptide in the pathophysiology of migraine and the levels of
plasma CGRP are increased in the external jugular during attacks in
migraine patients (12,13). The stimulation of the trigeminal
ganglion (TG) in animal models of migraine causes the release of
neuropeptides, including CGRP, substance P and neurokinin A, which
induces a series of peripheral and central events, including
vasodilatation (14), inflammation
and neuronal activation (15,16).
In addition to CGRP, cyclooxygenase-2 (COX-2) is an
important peripheral mediator of inflammation and pain. COX-2 can
increase prostaglandin E2 (PGE2) production
in the central nervous system and contribute to the severity of
pain responses in inflammatory pain (17,18).
Nonsteroidal anti-inflammatory drugs and selective inhibitors of
COX-2 (e.g. nimesulide; NM) have been used in migraine therapy for
decades, and can reduce plasma protein extravasation in
experimentally induced neurogenic inflammation of the rat dura
mater (19). This drug can also
attenuate c-Fos expression in the TNC of the electrical stimulation
of the trigeminal ganglion (ESTG) model (13).
Systemic administration of nitroglycerin (NTG), a
nitric oxide donor, can trigger a spontaneous-like migraine attack
in migraineurs, however, not in healthy individuals (20). In rats, subcutaneous administration
of NTG (10 mg/kg) can mimic a human migraine attack, which is the
closest possible simulation of the human NTG model (21,22).
Unilateral electrical stimulation of the trigeminal ganglion of
rats (UESTG) can induce structural alterations in CGRP positive
sensory nerve terminals and cause plasma protein leakage in the
dura mater (4). Therefore, UESTG
can induce chemical and vascular alterations that are similar to
those observed during a migraine attack.
The dura mater, TG and TNC are key parts of the TVS
and are essential in the process of inflammation and sensitization
in migraine (23). CGRP, p-ERK and
COX-2 are strongly associated with pain, particularly with the
transmission of nociceptive information. However, the function of
these three substances in neurogenic inflammation, central
sensitization and the intrinsic link among them during migraine
attacks has not been thoroughly examined.
The aim of the present study was to determine
whether p-ERK, CGRP and COX-2 are involved in migraine neurogenic
inflammation and central sensitization in the NTG-induced migraine
rat model. UESTG migraine model and NM were used to further assess
the possible functional connections between p-ERK, CGRP and COX-2
in migraine. Immunohistochemistry (IHC) was used to analyze the
protein expression of p-ERK, CGRP and COX-2.
Materials and methods
Animals
In total, 60 male Sprague-Dawley rats weighing
280–320 g (Vital River Laboratory Animal Technology Co., Ltd.,
Beijing, China) were used. All rats were kept under standard
laboratory housing conditions with a 12 h light-dark cycle and had
free access to food and water. All experimental protocols were
approved by the Ethics Committee for the Use of Experimental
Animals at Binzhou Medical University (Binzhou, China). All
procedures were undertaken with utmost caution to minimize the
suffering of animals. All rats were randomly divided into four
groups: Blank (n=6), NTG (n=36), ESTG (n=18) and NM (n=6) groups.
The NTG group (n=36) was then randomly divided into the NTG model
(n=18) and vehicle-treated (NS; n=18) groups. The ESTG group (n=18)
was also randomly divided into the ESTG model (n=6) and
sham-operation (SO; n=6) groups.
Experimental protocols
Administration of drugs
The rats in the NTG model group received weekly
subcutaneous (s.c.) injections of NTG (Beijing Yimin Pharmaceutical
Co., Ltd., Beijing, China) at a dose of 10 mg/kg for five
continuous weeks. For the control, the vehicle solution (0.9% NaCl)
was administered weekly via s.c. injections to the NS group rats
for five continuous weeks.
The rats in the NM group received intragastric
administration of the selective COX-2 inhibitor NM (Hainan Zhongrui
Kangzhi Pharmaceutical Co., Ltd., Hainan, China), which was
dissolved in saline in a volume of 10 ml/kg at a dose of 6
mg/kg/day for 7 days. Subsequently, 30 min after the last drug
administration, the rats were anesthetized with 10% chloral hydrate
(4 ml/kg; i.p.) and subjected to UESTG.
ESTG
Rats in the ESTG model group were anesthetized with
10% chloral hydrate (4 ml/kg, i.p.) and placed in a stereotaxic
frame (ZH-B; Zhenghua Biological Instrument Co., Ltd., Huaibei,
China). The calvarium was exposed by a midline incision. A hole was
drilled with a cranial drill 3.2–3.4 mm posterior to and 2.8–3.2 mm
laterally from the bregma. A disposable concentric needle electrode
(DCN37; Alpine Biomed Corp., Fountain Valley, CA, USA) was lowered
into the right TG (at a depth of ~9.2 mm from the dura mater). TG
was electrically stimulated for 30 min with square pulses at 10 Hz
and 0.5 mA with a pulse duration of 5 ms. Correct electrode
placement of the electrode needle was confirmed by ipsilateral
contraction of the masseter muscle during stimulation.
The rats in the SO group (n=6) underwent a surgical
procedure similar to that performed in the rats of the ESTG group.
However, the concentric bipolar electrode was only lowered into the
right TG and was maintained for only 30 min. The TG was not
electrically stimulated.
All rats in the NTG model and NS groups were
anesthetized with chloral hydrate (4 ml/kg, i.p.) at 30 min, 1 or 3
h after NTG or NS administration (n=6 each). The rats in the ESTG
model and SO groups were anesthetized for 30 min after stimulation
or sham-stimulation. Following being anesthetized, all rats were
transcardially perfused with 100–200 ml of 0.1 M phosphate-buffered
saline (PBS; pH 7.4; ZSGB-BIO, Beijing, China), followed by 500 ml
of cold and freshly made 4% paraformaldehyde (Tianjin No. 1 Organic
Chemical Plant, Tianjin,China) in 0.1 M PBS. Portions of the
cervical spinal cords, representing the lowest part of the TNC,
between 5 and 11 mm caudal to the obex were removed and postfixed
overnight for IHC. The ipsilateral dura mater and TG of the rats in
the ES model, SO and NM groups were also dissected and prepared for
IHC.
IHC
The dura mater, TG and TNC were fixed in 4% formalin
for at least 24 h, washed with 0.9% saline and processed with
ethanol and xylene solutions. The preparations were then embedded
in paraffin, cut into 4-µm thick sections and mounted on
glass slides following conventional procedures. The sections were
rinsed in PBS for 15 min and boiled in citrate buffer (pH 6.0) for
15 min for antigen retrieval. Following boiling, the sections were
immersed in methanol containing 0.3% H2O2 for
20 min. Sections were blocked with 1% bovine serum albumin at 21°C
for 10 min prior to incubation overnight at 4°C with one of the
following antibodies: Goat polyclonal anti-CGRP (1:300; cat. no.
ab36001; Abcam, Cambridge, MA, USA), rabbit polyclonal anti-COX-2
(1/350; cat. no. ab15191; Abcam) or mouse monoclonal anti-p-ERK
(1/50; cat. no. sc-7383; Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA). Following overnight incubation, preparations were
washed with PBS and incubated with MaxVision™ kits (Fuzhou Maixin
Biotech. Co., Ltd., Fuzhou, China), including monoclonal rabbit
anti-goat (Kit-5107), polyclonal goat anti-rabbit (Kit-5004) and
monoclonal goat anti-mouse (Kit-5001) immunoglobulin G secondary
antibodies, at 37°C for 30 min. Following incubation, the
preparations were washed thoroughly, incubated in
3,3′-diaminobenzidine tetrahydrochloride solution for color
detection and counterstained with hematoxylin.
Image acquisition and statistical
analysis
The immunolabeled specimens were examined under an
Olympus BX51 microscope (Olympus, Tokyo, Japan) equipped with a
DP72 camera (Olympus). Five images of each slide covered with
cultured cells were captured under x40 fixed magnification for the
TG and TNC and x100 for the dura mater. The measurement parameter
was the mean optical density (MOD) calculated using Image-Pro Plus
6.0 software (Media Cybernetics, Silver Spring, MD, USA).
All values are presented as the mean ± standard
deviation. Independent Student’s t-test was used to compare data
from two groups. One-way analysis of variance followed by Tukey’s
post-hoc test was applied when more than two groups of data were
compared. P<0.05 was considered to indicate a statistically
significant difference. GraphPad Prism 5.0 software (GraphPad Prism
Software Inc., San Diego, CA, USA) was used for statistical
analysis.
Results
Effect of NTG infusion on p-ERK, CGRP and
COX-2 protein expression in the dura mater, TG and TNC
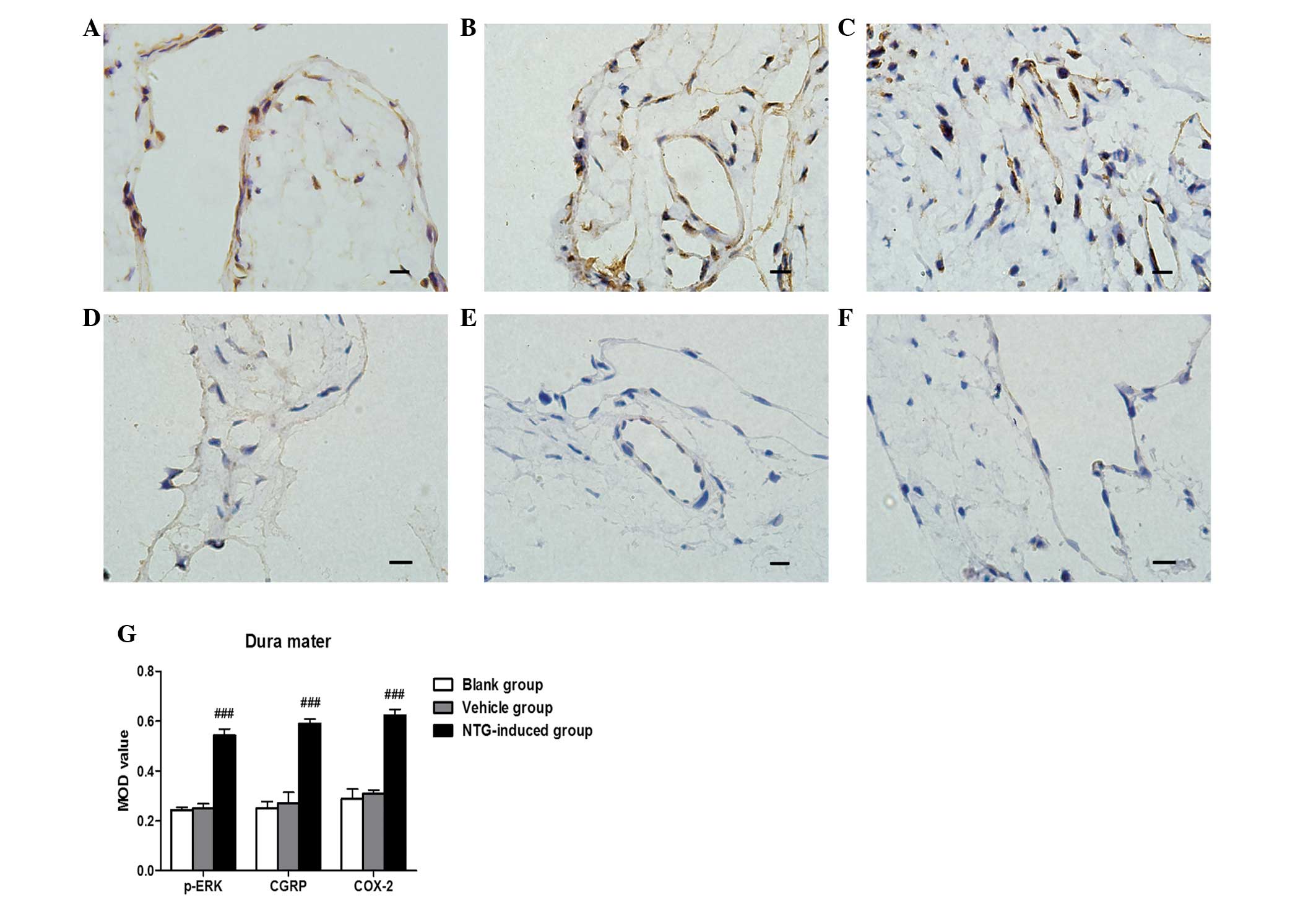
Based on IHC analysis, CGRP and COX-2 protein were
strongly expressed in the dura mater, TG and TNC 30 min, 1 or 3 h
after NTG infusion compared with the control rats (P<0.001;
Figs. 1Figure 2–3). No significant difference between CGRP
and COX-2 expression was observed 30 min, 1 or 3 h after NTG
infusion. For p-ERK, a temporal profile of NTG-induced
phosphorylation in the TVS was observed. Significantly higher
levels of p-ERK were found in the dura mater (F=72.72; P<0.01),
TG (F=68.08; P<0.01) and TNC (F=128.3; P<0.01) 30 min after
NTG administration compared with the controls. The p-ERK levels
gradually decreased and were close to the basal level by 3 h
(Fig. 4). Vehicle (NS)-treated
rats demonstrated low basal levels of p-ERK, CGRP and COX-2 protein
expression in the dura mater, TG and TNC at 30 min, 1 and 3 h after
vehicle infusion (Figs. 1Figure 2–3). Furthermore, no significant difference
in the expression of p-ERK, CGRP and COX-2 was observed at 30 min,
1 or 3 h in vehicle (NS)-treated rats.
 | Figure 2Representative images of p-ERK, CGRP
and COX-2 immunoreactivity in the TG 30 min after (A–C) NTG or
(D–F) vehicle infusion by immunohistochemistry and (G) analysis of
the MOD of p-ERK, CGRP and COX-2 expression in the TG. An increase
in (A) p-ERK, (B) CGRP and (C) COX-2 expression was observed in the
TG following NTG infusion and the MOD value for NTG-treated rats
was significantly higher than in the vehicle and blank groups.
(###P<0.001, compared with the vehicle and blank
groups; n=6 in each group; error bars indicate standard deviation;
scale bar=100 µm). MOD, mean optical density; p-ERK, phosphorylated
extracellular signal-regulated kinase; CGRP, calcitonin
gene-related peptide; COX-2, cyclooxygenase-2; TG, trigeminal
ganglion; NTG, nitroglycerin. |
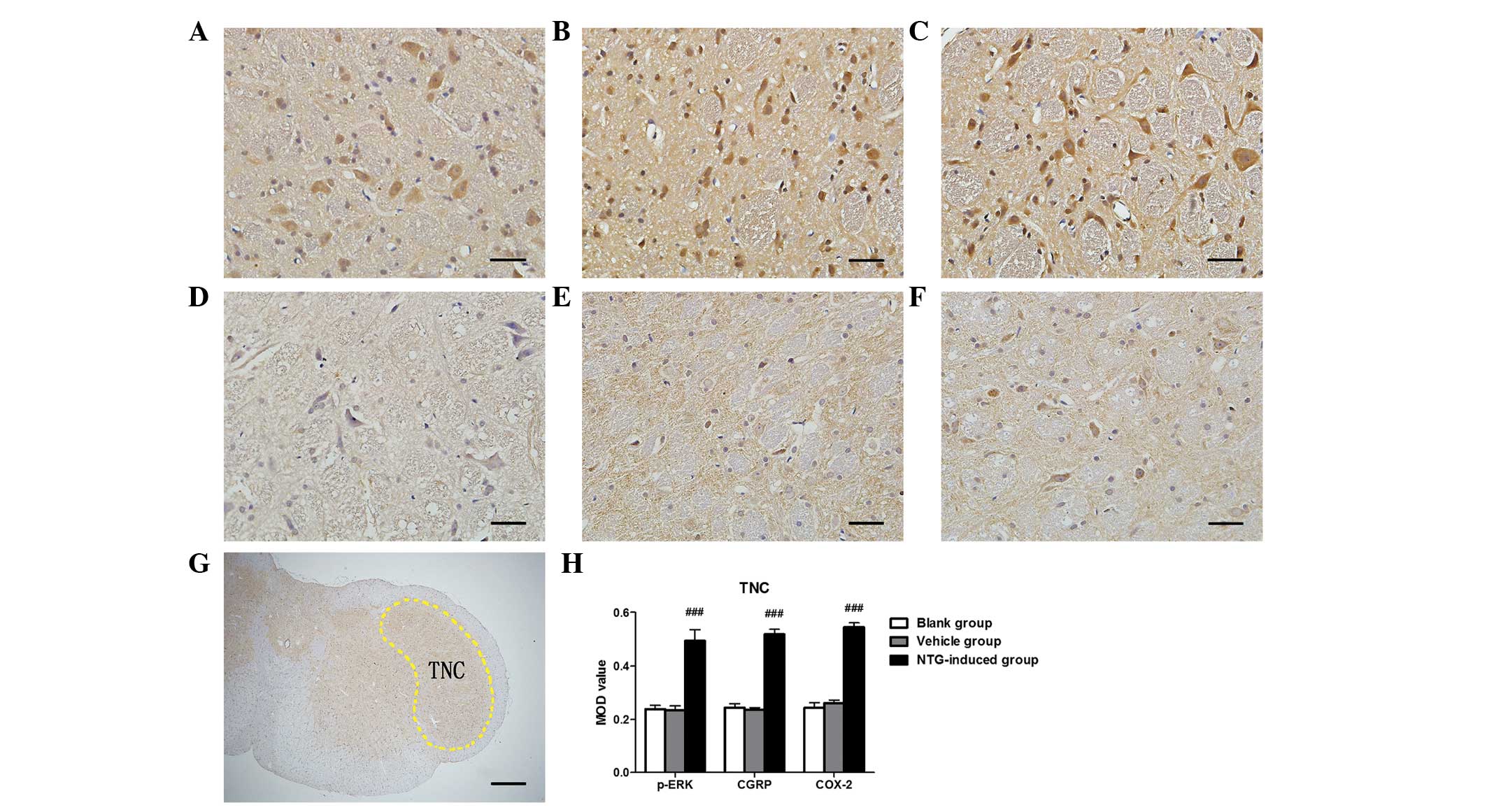 | Figure 3Representative images of p-ERK, CGRP
and COX-2 immunoreactivity in the TNC 30 min after (A–C) NTG or
(D–F) vehicle infusion by immunohistochemistry, (G) rat TNC section
nuclei and (H) analysis of the MOD of p-ERK, CGRP and COX-2
expression in the TG. An increase in (A) p-ERK, (B) CGRP and (C)
COX-2 expression was observed in the TNC following NTG infusion and
the MOD value for NTG-treated rats was significantly higher than in
the vehicle and blank groups. [###P<0.001, compared
with the vehicle and blank groups; n=6 in each group; error bars
indicate standard deviation; scale bar (A–F)=100 µm; scale
bar (G)=1 mm]. MOD, mean optical density; p-ERK, phosphorylated
extracellular signal-regulated kinase; CGRP, calcitonin
gene-related peptide; COX-2, cyclooxygenase-2; TNC, trigeminal
nucleus caudalis; NTG, nitroglycerin. |
 | Figure 4Expression of p-ERK in the (A) dura
mater, (B) TG and (C) TNC following NTG infusion. As shown in the
histogram, the phosphorylation of ERK following NTG infusion of the
dura mater, TG and TNC demonstrated a temporal profile.
Significantly higher levels of p-ERK were found in the dura mater,
TG and TNC 30 min after NTG administration compared with the
controls. The p-ERK levels gradually decreased and were close to
the basal level by 3 h. (**P<0.01, compared with the
NS group; n=6 in each group; error bars indicate standard
deviation). p-ERK, phosphorylated extracellular signal-regulated
kinase; TNC, trigeminal nucleus caudalis; TG, trigeminal ganglion;
NTG, nitroglycerin; NS, vehicle-treated rats. |
Effect of electrical stimulation and
pretreatment with NM on p-ERK, CGRP and COX-2 protein expression in
the dura mater, TG and TNC
The surgical procedure and lowering of the electrode
into the TG did not significantly increase the expression of p-ERK,
CGRP and COX-2 in the TNC, ipsilateral side of the dura mater or
the TG. Following electrical stimulation, a significant increase in
p-ERK, CGRP and COX-2 was observed in the TNC, ipsilateral dura
mater and TG compared with the sham-surgery group (P<0.001).
Pretreatment with NM (6 mg/kg/day for 7 days) resulted in a
significant decrease in p-ERK, CGRP and COX-2 MOD values in the
TNC, ipsilateral dura mater and TG compared with the
electrically-stimulated rats (P<0.05). Pretreatment with NM also
demonstrated a significant increase in p-ERK, CGRP and COX-2 in the
TNC, ipsilateral dura mater and TG compared with the sham-surgery
group (P<0.001) and blank control group (P<0.001). No
differences were detected between the sham-surgery and the blank
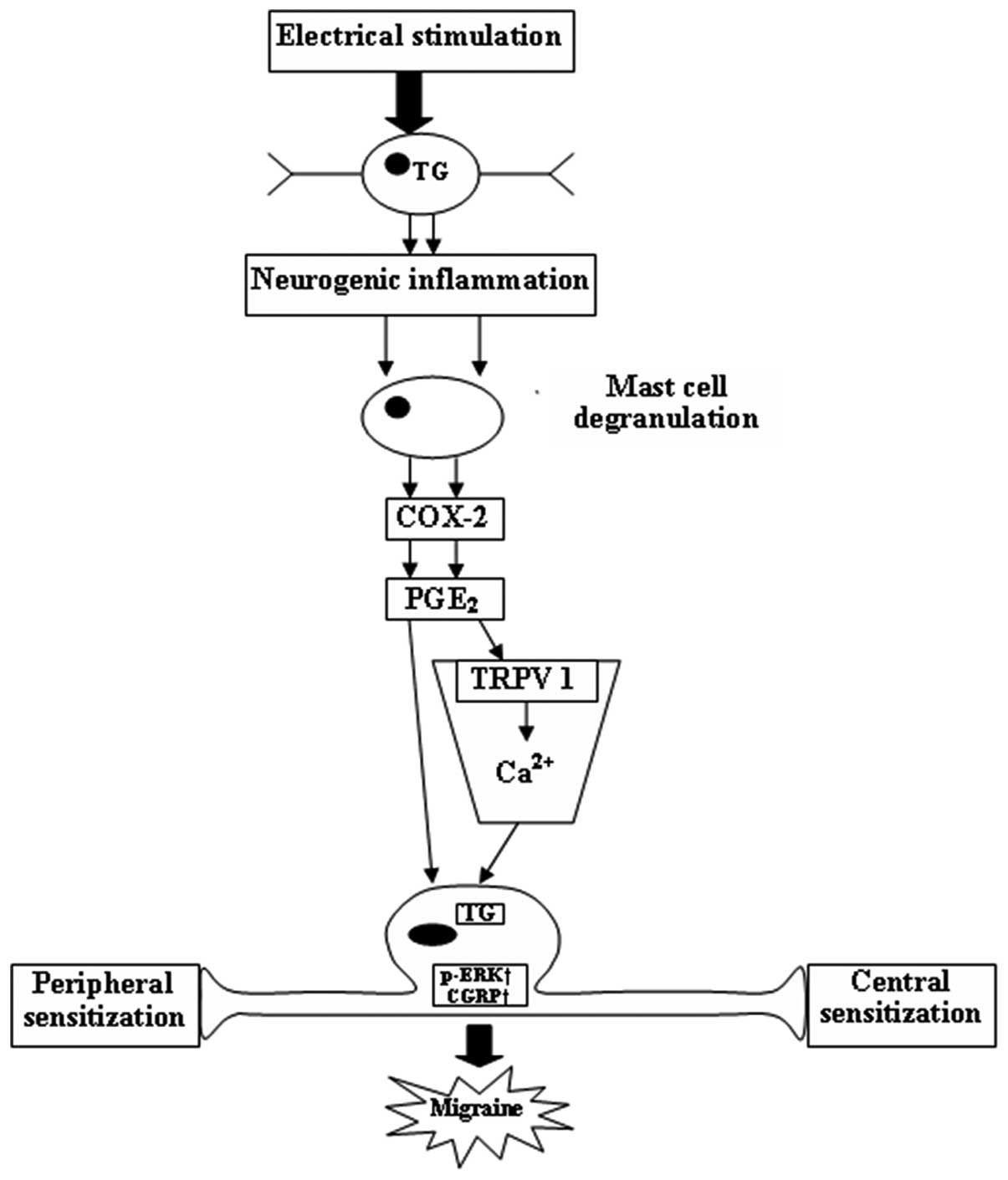
control (Fig. 5). A schematic
diagram shows the connections between p-ERK, CGRP and COX-2 in the
pathophysiological mechanisms of migraine (Fig. 6).
 | Figure 5Effect of electrical stimulation and
pretreatment with NM on the protein expression of p-ERK, CGRP and
COX-2 in the (A) dura mater, (B) TG and (C) TNC. As shown in the
histogram, following electrical stimulation, a significant increase
in p-ERK, CGRP and COX-2 was observed in the TNC, ipsilateral dura
mater and TG compared with the sham-surgery and blank groups.
Pretreatment with NM demonstrated a significant decrease in p-ERK,
CGRP and COX-2 MOD values in the TNC, ipsilateral dura mater and TG
compared with the electrically-stimulated rats, however this was
higher than in the sham-surgery and blank groups. No differences
were detected between the sham-surgery and the blank groups.
(*P<0.05, compared with the electrically-stimulated
group; ###P<0.001, compared with the sham-surgery and
blank groups; n=6 in each group; error bars indicate standard
deviation). MOD, mean optical density; p-ERK, phosphorylated
extracellular signal-regulated kinase; CGRP, calcitonin
gene-related peptide; COX-2, cyclooxygenase-2; NM, nimesulide;
ESTG, electrical stimulation of the trigeminal ganglion; SO, sham
operation; TNC, trigeminal nucleus caudalis; TG, trigeminal
ganglion. |
Discussion
The present study demonstrated that infusion of NTG
and ESTG upregulated p-ERK, CGRP and COX-2 protein expression
within the dura mater, TG and TNC of rats. A temporal profile of
NTG-induced p-ERK was observed in the TVS. NM, a selective COX-2
inhibitor, attenuated the expression of p-ERK, CGRP and COX-2
following ESTG in rats.
Numerous hypotheses regarding the pathophysiology of
migraine exist. The generally accepted neurovascular theory states
that migraines are mediated by prolonged activation of meningeal
nociceptors, which are located in the dura mater and vessels
(1,23). The neurovascular theory centers on
the activation of the TVS. The TVS consists of pseudounipolar
neurons in the TG that has first-order afferent neurons innervating
the pial and dural meningeal vessels, and efferent projections
synapsing with second-order neurons in the TNC, which provides
projections to several higher brain centers, including the
posterior thalamus, hypothalamus and cortex (2,23).
Activation of perivascular trigeminal nerves within meninges causes
the release of CGRP, substance P and neurokinin A, which leads to a
series of peripheral and central events, including inflammation and
peripheral/central sensitization (24).
Central sensitization is the process that underlies
migraine-associated allodynia (25). Allodynia is a state in which
trigeminal neurons are elicited by persistent pain through
activation of meningeal perivascular pain fibers and second-order
brainstem trigeminal neurons (5,25).
Consequently, meningeal perivascular pain fibers become
hyper-responsive to all subsequent stimuli delivered to the
receptive fields of neurons (2,6).
Cutaneous allodynia, which has been found to be more common in
chronic migraineurs, reinforces the hypothesis stating the
necessity of frequent stimulation of central nuclei of the pain
pathway to induce sensitization (7). Based on clinical symptoms, the
pathophysiology of migraine can be divided into three phases: The
trigger phase characterized by neuronal hyperexcitability, the aura
phase involving cortical spreading depression (CSD) and the
headache phase precipitated by activation and sensitization of the
TVS (26,27). Central sensitization is important
in the headache stages of migraine attacks and introduces the brain
into a state of excessive sensitivity (2).
In rats, administration of NTG activates
second-order nociceptors in the TNC and produces an increased level
of nitric oxide synthase in the area associated with the central
sensitization phenomenon-hyperalgesia/cutaneous allodynia (22,28).
The NTG dose (10 mg/kg, for five continuous weeks, s.c.) selected
for the present study was used to build a migraine hyperalgesia
model that mimics chronic migraineurs. The model has provided
interesting insights into the neuropharmacological mechanisms of
the initiation and recurrence of migraine attacks (21,29).
Thus, the present study reported a significant increase in p-ERK,
CGRP and COX-2 expression in the dura mater, TG and TNC, which are
the three key structures in TVS for migraine genesis following NTG
administration. These results suggest that the activation of p-ERK,
CGRP and COX-2 is crucial in neurogenic inflammation and central
sensitization of migraine.
In addition, an interesting time-dependent mechanism
for p-ERK synthesis resulting from NTG infusion was demonstrated.
Numerous studies have demonstrated that the nociceptive stimulation
of peripheral C-fibers could induce p-ERK in the dorsal root
ganglion (DRG) (9,30). The induction p-ERK may be
associated with the hypersensitivity of spinal neurons in
inflammatory pain. Dai et al observed a transient
upregulation of p-ERK minutes following TRPV1 stimulation in the
DRG. However, the levels of p-ERK returned to baseline after 120
min (30). The present study
demonstrated a similar time course in terms of the upregulation of
p-ERK. The present study also found a transient upregulation of
p-ERK in the dura mater, TG and TNC, 30 min after NTG infusion. The
levels of p-ERK then gradually decreased to baseline levels before
180 min. According to our experimental observations, the
NTG-induced phenomenon in rats that mimic migraine attack in
migraineurs, which are characterized by continuous head scratching
and climbing behavior, did not appear until 20–30 min after NTG
administration. Based on the three phases of migraine
pathophysiology, it was hypothesized that p-ERK primarily functions
during the early onset of a migraine attack, which involves
neuronal hyperexcitability and CSD. A clear time course expression
of CGRP and COX-2 was not observed in TVS, suggesting that CGRP and
COX-2 function in central sensitization during the headache phase
of migraine pathophysiology, which maintained a migraine attack for
a relatively long time period in the NTG infusion model.
ESTG, resulting in the release of CGRP, which was
involved in inflammation and nociceptive information transmission,
has been used to mimic neurogenic inflammation and examine migraine
pathophysiology (27). ESTG has a
direct effect on first-order sensory neurons, thereby causing
alterations in the peripheral endings to release mediators from
perivascular trigeminal nerves within meninges, which results in
neurogenic inflammation (3). In
the central endings, a marked activation of second-order neurons in
the TNC is observed. In the present study, the ESTG models were
used for a more in-depth examination of the possible functional
connections between p-ERK, CGRP and COX-2 in migraine mechanisms. A
significant increase in p-ERK, CGRP and COX-2 was found in the dura
mater, TG and TNC of ESTG-induced rats, suggesting that the
nociceptive stimulation in the TG activated the synthesis of p-ERK,
CGRP and COX-2 in peripheral and central areas of the TVS, which
are important in neurogenic inflammation and central sensitization
during migraine. NM attenuated the expression of ESTG-induced p-ERK
and CGRP in rat TVS structures. COX-2 may have stimulated the
production of p-ERK and CGRP. The synthesis of p-ERK and CGRP was
inhibited by the COX-2 inhibitor. Our findings are in agreement
with the results obtained by Neeb et al who demonstrated
that the activation of neuronal cells in the TG by interleukin-1β
can lead to an elevated expression of COX-2 and that newly
synthesized PGE2 (by COX-2) activates trigeminal neurons
to release CGRP (16). Findings
from this study support the assumption that a sequential link
between COX-2 and CGRP exists. The present study also observed a
downregulated expression of p-ERK in the TVS of NM-induced ESTG
rats. Iwashita et al indicated that CSD can activate dural
TRPV1 to send nociceptive signals to the TVS by facilitating
degranulation of mast cells in the dura mater (11). PGE2, serotonin and
histamine, released by mast cells, are known to induce TRPV1
sensitization (11,31). The influx of Ca2+ via
TRPV1 upregulated the level of p-ERK and caused peripheral
hypersensitivity via transcriptional regulation (32). Thus, COX-2, which synthesizes
PGE2, can increase the synthesis of p-ERK by inducing
TRPV1 sensitization. COX-2 can also transmit nociceptive signals to
the peripheral and central area (11).
Thus, p-ERK, CGRP and COX-2 may function in
neurogenic inflammation and central sensitization, which are
relevant in migraine modulation. It was also found that p-ERK may
be involved in the pathogenesis of an early onset migraine attack.
In addition, the attenuation of p-ERK and CGRP release could
contribute to the effect of COX-2 inhibitors, which hinder
sensitization and alleviate pain. CGRP and p-ERK may improve our
understanding of the mechanisms of COX-2 inhibitors in migraine
therapy.
Acknowledgments
This study was supported by the Science and
Technology Development Project in Binzhou (project no.
2011ZC0908).
References
|
1
|
Noseda R and Burstein R: Migraine
pathophysiology: Anatomy of the trigeminovascular pathway and
associated neurological symptoms, cortical spreading depression,
sensitization and modulation of pain. Pain. 154(Suppl 1): S44–S53.
2013. View Article : Google Scholar
|
|
2
|
Kojić Z and Stojanović D: Pathophysiology
of migraine - from molecular to personalized medicine. Med Pregl.
66:53–57. 2013. View Article : Google Scholar
|
|
3
|
Goadsby PJ: Recent advances in
understanding migraine mechanisms, molecules and therapeutics.
Trends Mol Med. 13:39–44. 2007. View Article : Google Scholar
|
|
4
|
Knyihar-Csillik E, Tajti J, Mohtasham S,
Sari G and Vecsei L: Electrical stimulation of the Gasserian
ganglion induces structural alterations of calcitonin gene-related
peptide-immunoreactive perivascular sensory nerve terminals in the
rat cerebral dura mater: a possible model of migraine headache.
Neurosci Lett. 184:189–192. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Louter MA, Bosker JE, van Oosterhout WP,
et al: Cutaneous allodynia as a predictor of migraine
chronification. Brain. 136:3489–3496. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Burstein R, Yarnitsky D, Goor-Aryeh I,
Ransil BJ and Bajwa ZH: An association between migraine and
cutaneous allodynia. Ann Neurol. 47:614–624. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lovati C, Mariotti C, Giani L, et al:
Central sensitization in photophobic and non-photophobic
migraineurs: possible role of retino nuclear way in the central
sensitization process. Neurol Sci. 34(Suppl 1): S133–S135. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Roux PP and Blenis J: ERK and p38
MAPK-activated protein kinases: a family of protein kinases with
diverse biological functions. Microbiol Mol Biol Rev. 68:320–344.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shimizu K, Asano M, Kitagawa J, et al:
Phosphorylation of extracellular signal-regulated kinase in
medullary and upper cervical cord neurons following noxious tooth
pulp stimulation. Brain Res. 1072:99–109. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang X, Kainz V, Zhao J, Strassman AM and
Levy D: Vascular extracellular signal-regulated kinase mediates
migraine-related sensitization of meningeal nociceptors. Ann
Neurol. 73:741–750. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Iwashita T, Shimizu T, Shibata M, et al:
Activation of extracellular signal-regulated kinase in the
trigeminal ganglion following both treatment of the dura mater with
capsaicin and cortical spreading depression. Neurosci Res.
77:110–119. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Durham PL: Calcitonin gene-related peptide
(CGRP) and migraine. Headache. 46(Suppl 1): S3–S8. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim GM, Jin KS and Chung CS: Differential
effects of corticosteroids on the expression of cyclooxygenase-2,
tumour necrosis factor-alpha and matrix metalloproteinase-9 in an
animal model of migraine. Cephalalgia. 28:1179–1187. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Waeber C and Moskowitz MA: Migraine as an
inflammatory disorder. Neurology. 64(Suppl 2): S9–S15. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Storer RJ, Akerman S and Goadsby PJ:
Calcitonin gene-related peptide (CGRP) modulates nociceptive
trigeminovascular transmission in the cat. Br J Pharmacol.
142:1171–1181. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Neeb L, Hellen P, Boehnke C, et al: IL-1β
stimulates COX-2 dependent PGE2 synthesis and CGRP
release in rat trigeminal ganglia cells. PLoS one. 6:e173602011.
View Article : Google Scholar
|
|
17
|
Kawabata A: Prostaglandin E2 and pain - an
update. Biol Pharm Bull. 34:1170–1173. 2011. View Article : Google Scholar
|
|
18
|
Tassorelli C, Greco R, Armentero MT,
Blandini F, Sandrini G and Nappi G: A role for brain
cyclooxygenase-2 and prostaglandin-E2 in migraine: effects of
nitroglycerin. Int Rev Neurobiol. 82:373–382. 2007.PubMed/NCBI
|
|
19
|
Varga H, Pardutz A, Vamos E, et al: Cox-2
inhibitor attenuates NO-induced nNOS in rat caudal trigeminal
nucleus. Headache. 47:1319–1325. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Iversen HK, Olesen J and Tfelt-Hansen P:
Intravenous nitroglycerin as an experimental model of vascular
headache. Basic characteristics. Pain. 38:17–24. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tassorelli C and Joseph SA: Systemic
nitroglycerin induces Fos immunoreactivity in brainstem and
forebrain structures of the rat. Brain Res. 682:167–181. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tassorelli C, Greco R, Wang D, Sandrini M,
Sandrini G and Nappi G: Nitroglycerin induces hyperalgesia in rats
- a time-course study. Eur J Pharmacol. 464:159–162. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kaiser EA and Russo AF: CGRP and migraine:
Could PACAP play a role too? Neuropeptides. 47:451–461. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ramachandran R, Bhatt DK, Ploug KB, et al:
Nitric oxide synthase, calcitonin gene-related peptide and NK-1
receptor mechanisms are involved in GTN-induced neuronal
activation. Cephalalgia. 34:136–147. 2014. View Article : Google Scholar
|
|
25
|
Aguggia M, Saracco M, Cavallini M, Bussone
G and Cortelli P: Sensitization and pain. Neurol Sci. 34(Suppl 1):
S37–S40. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Silberstein SD: Migraine pathophysiology
and its clinical implications. Cephalalgia. 24(Suppl 2): 2–7. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Arulmani U, Gupta S, VanDenBrink AM,
Centurión D, Villalón C and Saxena P: Experimental migraine models
and their relevance in migraine therapy. Cephalalgia. 26:642–659.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Varga H, Pardutz A, Vamos E, et al:
Selective inhibition of cyclooxygenase-2 attenuates
nitroglycerin-induced calmodulin-dependent protein kinase II alpha
in rat trigeminal nucleus caudalis. Neurosci Lett. 451:170–173.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tassorelli C, Greco R, Morazzoni P, Riva
A, Sandrini G and Nappi G: Parthenolide is the component of
tanacetum parthenium that inhibits nitroglycerin-induced Fos
activation: studies in an animal model of migraine. Cephalalgia.
25:612–621. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dai Y, Iwata K, Fukuoka T, et al:
Phosphorylation of extracellular signal-regulated kinase in primary
afferent neurons by noxious stimuli and its involvement in
peripheral sensitization. J Neurosci. 22:7737–7745. 2002.PubMed/NCBI
|
|
31
|
Levy D, Burstein R, Kainz V, Jakubowski M
and Strassman AM: Mast cell degranulation activates a pain pathway
underlying migraine headache. Pain. 130:166–176. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Obata K and Noguchi K: MAPK activation in
nociceptive neurons and pain hypersensitivity. Life Sci.
74:2643–2653. 2004. View Article : Google Scholar : PubMed/NCBI
|