Introduction
Leber’s hereditary optic neuropathy (LHON; Online
Mendelian Inheritance in Man #535000) is characterized by the
selective degeneration of retinal ganglion cells, particularly
contributing to the papillomacular bundle, which leads to optic
atrophy and loss of central vision (1). Typically, the clinical presentation
includes acute or subacute central visual loss in one eye, with
effects in the other eye 2–4 months later (2), although symptoms occur in the two
eyes simultaneously in 25% of patients (3). The disease was first described by
Leber in 1871 (4), Wallace et
al reported the mitochondrial DNA (mtDNA) G11778A point
mutation in patients with LHON in 1988 (5). In total >95% of LHON pedigrees are
known to exhibit one of three major mtDNA point causative
mutations: G3460A (13% of cases), G11778A (69% of cases) and
T14484C (14% of cases) (6), which
are located in the ND1, ND4 and ND6 mtDNA genes, respectively, and
affect the genes encoding complex I subunits of the mitochondrial
respiratory chain, which lead to disorders of the oxidative
phosphorylation system (7,8). However, the three major mutations are
only responsible for 38.3% cases in Chinese individuals with LHON
(9) and a number of other
pathogenic mtDNA variants have been reported, which are awaiting
confirmation for LHON pathogenicity (10).
At present, two key features of LHON require
clarification. There is a marked incomplete penetrance and gender
bias, with only 50% of male and 10% of female carriers eventually
losing their vision (11). This
indicates that there are additional genetic or environmental
factors in the pathophysiology of the disorders. From then on, the
importance of the mitochondrial haplogroup has been considered. A
haplogroup is a collection of polymorphisms, which reflect the
evolutionary history of the mtDNA molecule (12). In western European, the J2 and J1
haplogroups have been reported to contribute to increased risk of
visual failure in families with G11778A and G3460A mutations,
respectively (13). In Chilean
patients, the Amerindian haplogroup A2 has been associated with
delayed onset of LHON and haplogroup C has been associated with
improved vision (14). In a
previous study, which investigated Chinese patients of G11778A
haplogroup F, a protective effect against LHON was observed, whilst
haplogroups M7b and M10a were identified as risk factors (15,16).
The present study aimed to detect LHON-associated mutations and
mitochondrial haplotypes in two families, and investigate the
effects of the interaction between mutations and mitochondrial
haplotypes on the phenotypic manifestation of LHON.
Materials and methods
Participants
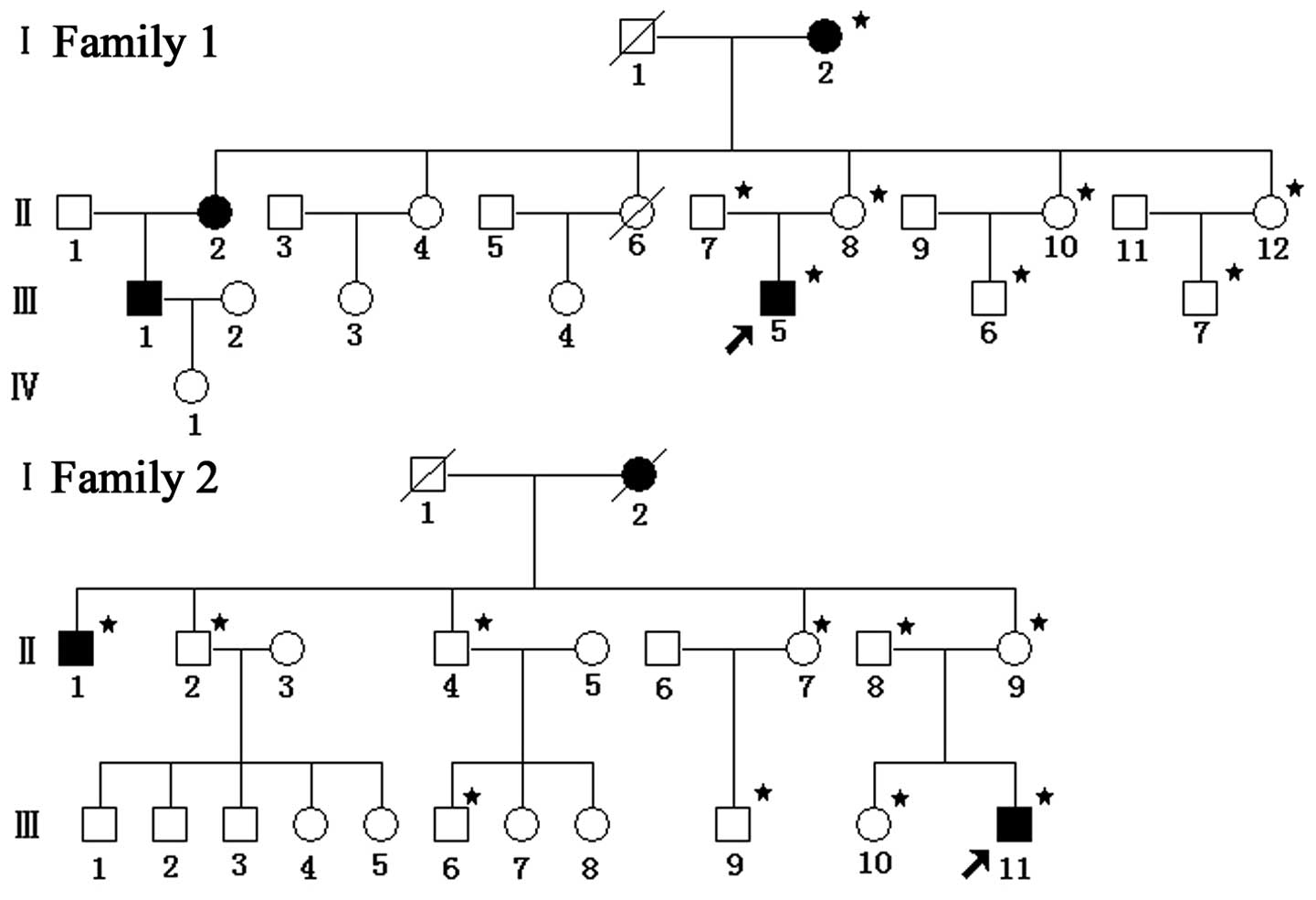
Individuals from two unrelated families, each
containing 22 members, were selected, and certain family members
(marked with an asterisk in Fig.
1) were enrolled in the present study. Prior to involvement in
the clinical evaluations, informed consent and blood samples were
obtained from all the family members involved. Also, informed
consent was obtained from family members for minors involved in
this study. The present study was approved by the Ethics Committee
of Zhongnan Hospital of Wuhan University (Wuhan, China) and was in
line with the Declaration of Helsinki. Personal and family medical
histories, history of tobacco use and alcohol intake, and details
of any other clinical abnormalities were also obtained.
Ophthalmologic evaluations
The ophthalmologic examinations of all patients and
available relatives were performed in the department of
Ophthalmology at Zhongnan Hospital of Wuhan University (Wuhan,
China). The examinations included measuring visual acuity, fundus
examination, visual field examination, visual evoked potentials
(VEP), fundus photography (TRC-50EX Fundus; Topcon Itabashi-ku
Corporation, Tokyo, Japan), and head, orbital and cervical spine
magnetic resonance imaging (MRI; Magnetom Trio Tim 3.0 MRI;
Siemens, Munich, Germany). The level of visual impairment was
defined according to the visual acuity, as follows: Normal, ≥0.3;
0.1≤ mild <0.3; 0.05≤ moderate <0.1; 0.02≤ severe <0.05;
and profound, <0.02.
Mutational analysis
Venipuncture was performed on all the family members
participated, with the exception of family 1-I-2. Approximately 2
ml EDTA-anticoagulant peripheral blood was drawn from participants
for DNA collection. Approximately 0.5 ml blood and 3X Red Blood
Cell Lysis Buffer (1 mM, 1.5 ml) were adding to a 10-ml EP tube.
The mixture was placed in an ice bath for 30 min prior to
centrifugation at 13,700 × g for 10 min at 4°C (TGL-18R; Hema
Medical Instruments Co., Ltd., Zhuhai, China). The upper aqueous
component was removed and 3X nucleus pyrolysis liquid (1mM, 1.5
ml), 150 μl of 10% sodium dodecyl sulphate (SDS) and 15
μl of 10 mg/ml proteinase K (Sigma-Aldrich, St. Louis, MO,
USA) were added, and the mixture was put into the Oven Controlled
Crystal Oscillator (IS RSV1; Crystal Technology and Industries,
Inc., Dallas, TX, USA) at 37°C and 200 × g overnight. Then the
digestion solution was gently mixed with equal volume of
Tris-phenol (2 mM, pH 8.0). The phases were separated by
centrifugation at 13,700 × g for 10 min at 4°C. The upper aqueous
component was retained and equal volume of Phenol/chloroform
(volume, 1:1) was added, then the mixture was centrifuged at 13,700
× g for 10 min at 4°C. The majority of the aqueous phase was
transferred into a 1.5 ml EP tube prior to mixing with 1 ml of 70%
ethanol. Later the phases were separated by centrifugation at room
temperature at 13,700 × g for 10 min twice. The aqueous phase was
decanted and the precipitation contained the desired DNA. After
draining excess liquid, the precipitation was dissolved with 50
μl TE and stored at 4°C. While the genomic DNA of family
1-I-2 was extracted using a Chelex-100 (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) method from the scapus pili. The mtDNA fragments
of ND1, ND4 and ND6, which contain three major mutations (G3460A,
G11778A and T14484C), were amplified from the DNA extracted from
all family members involved. The PCR products were directly
sequenced using one of the PCR primers. The sequences were blasted
with the reference sequences to identify the mutations. The
amplification of the entire mtDNA of the two probands were
performed using previously defined 24 primers, covering overlapped
fragments, using polymerase chain reaction (PCR) (17). Each amplification reaction
contained 5.0 μl of 10X Ex Taq Buffer, 5.0 μl
MgCl2 (25 mM), 2.0 μl of 4X dNTP (2mM), 2.0
μl of 1 mM each primer, 2 units of Taq polymerase (Thermo
Fisher Scientific, Inc., Waltham, MA, USA), 3.0 μl genomic
DNA and double distilled water was added to reach 50 ml reaction
volume. The protocol for amplification reactions was as follows:
Denaturation at 95°C for 5 min followed by 35 cycles of a
denaturation at 94°C for 30 sec, an annealing at 52–63°C for 30 sec
and an extension at 72°C for 45 sec, the terminal extension step at
72°C for 10 min. The amplification was conducted using a Hema 9600
PCR thermocycler (Hema Medical Instruments Co., Ltd.) and a 3130xl
Genetic Analyzer (Applied Biosystems Life Technologies, Carlsbad,
CA, USA). The bidirectional sequence results were analyzed and
compared with the updated consensus revised Cambridge reference
sequence (GenBank accession no. NC_012920). Changes in the mtDNA
were compared against a mitochondrial database, including Mitomap
(www.mitomap.org/MITOMAP) and mtDB
(www.mtdb.igp.uu.se/).
Phylogenetic analysis
Conservative amino acid analyses of missense
mutations were identified in the Mitochondrial Single Nucleotide
Polymorphism public database (http://mtsnp.tmig.or.jp/mtsnp/index_e.shtml), which
compared the human amino acid variants with those of 60
vertebrates, including Artibeus jamaicensis, Didelphis
virginiana and Hippopotamus amphibius. The degree of
conservation is defined as the percentage of species in the 60
vertebrates with the same amino acid in the same position as in
humans (18).
Haplogroup analyses
The entire mtDNA sequence of the probands was
assigned to mitochondrial haplogroups using the nomenclature of the
mitochondrial haplogroups (19),
according to PhyloTree (http://www.phylotree.org/) (20).
Results
In family 1, the proband (III-5) was a 17 year old
male from Henan, China. This individual developed impaired vision
in the right eye, followed four days later by impaired vision in
the left eye, with no clear cause. As presented in Table I, III-5 presented with 0.05 visual
acuity in the right eye and 0.01 visual acuity in the left eye,
with dense central scotomas and optic disk pallor bilaterally
(21). Visual field analysis
revealed large visual field defects connected to the physiological
blind spot in the right and left eye. The flash VEP demonstrated
bilateral delays in the amplitudes of the latency. No other
clinical abnormalities were observed on the head, orbital and
cervical spine MRI. In addition, III-5 had a history of tobacco and
alcohol use for almost two years. In the matrilineal relatives,
there were 2 affected males and 2 affected females; thus, the ratio
of affected males to females was 1:1. However, in total there were
4 males and 9 females in this family. Thus, the morbidity of males
and females was 2/4 and 2/9, respectively. The ratio of 2/4 to 2/9
was the incidence between the males and females, which equaled
2.25:1, and the penetrance of visual loss (affected matrilineal
relatives/total matrilineal relatives) was 30.8%.
 | Table IClinical characteristics of patients
carrying the G11778A mutation. |
Table I
Clinical characteristics of patients
carrying the G11778A mutation.
| Patient | Age
(years)/gender | Onset age
(years) | Visual acuity
| Level of visual
impairment |
|---|
| Right eye | Left eye |
|---|
| Family 1-I-2 | 75/M | N/A | 0.04 | 0.06 | Severe |
| Family 1-III-5 | 17/F | 17 | 0.05 | 0.01 | Severe |
| Family 2-II-1 | 57/F | 17 | 0.01 | HM | Profound |
| Family 2-III-11 | 12/F | 12 | 0.05 | 0.02 | Severe |
In family 2, the proband (III-11) was a 12 year old
male from Hubei, China, who exhibited painless, progressive
deterioration of the left eye, followed by rapid vision loss in his
right eye 11 months later. Visual acuity was 0.05 and 0.02 in the
right and left eyes, respectively. No other clinical abnormalities
of the eye were observed. The ratio between the number of affected
males and females was 2:1 and the incidence in males and females
was 2/5 and 1/4 respectively. Therefore, the males:female incidence
ratio was 1.6:1 and the penetrance of visual loss was 33.3%.
In order to elucidate the molecular basis of LHON,
mutational screening of three major mutations, G3460A, G11778A and
T14484C, was performed in the two families using PCR direct
bidirectional sequencing. The results revealed the presence of
G11778A in the two families, but absence of the G3460A and T14484C
mutations.
To determine the role of the haplogroup in the
phenotypic expression of the G11778A mutation, the entire
mitochondrial genome was analyzed using PCR direct sequencing, as
shown in Table II. These missense
variants were further evaluated by phylogenetic analysis among 60
types of vertebrate, particularly the T3394C and T14502C mutations,
which are considered to be secondary mutations in the expression of
LHON (18,21,22).
The analyses revealed that T3394C was highly conserved in 58/60
mammalian species and the conservative degree was 96.7%. G11778A
was highly conserved in 59/60 mammalian species, with a
conservative degree of 98.3%. The degree of conservation of T14502C
was 72.13%, which suggested it as a potential polymorphism. The
analyses of the complete mtDNA sequences indicated that the two
families exhibited the H2a2a1 haplogroup (Fig. 2).
| Table IIList of the mtDNA variants identified
in the two families containing individuals diagnosed with Leber’s
hereditary optic neuropathy. |
Table II
List of the mtDNA variants identified
in the two families containing individuals diagnosed with Leber’s
hereditary optic neuropathy.
| Gene | Position | Replacement | CRS | Family 1 | Family 2 | Conservative
degreea (%) |
|---|
| D loop | 73 | A>G | A | G | G | |
| 146 | T>C | T | C | C | |
| 152 | T>C | T | C | C | |
| 204 | T>C | T | – | C | |
| 263 | A>G | A | G | G | |
| 272 | A>G | A | – | G | |
| 489 | A>G | T | – | C | |
| 16093 | T>C | T | C | C | |
| 16193 | C>T | C | T | T | |
| 16223 | C>T | C | T | T | |
| 16311 | T>C | T | – | C | |
| 16357 | T>C | T | C | C | |
| 16497 | A>G | A | G | G | |
| 16519 | T>C | T | C | – | |
| 12SrRNA | 709 | G>A | G | A | A | |
| 750 | A>G | A | G | G | |
| 1438 | A>G | A | G | G | |
| 16SrRNA | 2706 | A>G | A | G | G | |
| NDI | 3394 | T>C
(Ty83His) | T | C | – | 96.7 |
| 4140 | C>T | C | T | T | |
| ND2 | 4769 | A>G | A | G | G | |
| COI | 6965 | T>G | T | G | – | |
| 7028 | C>T | C | T | T | |
| 7250 | A>G | A | G | G | |
| ATP6 | 8701 | A>G
(Thr59Ala) | A | G | G | 28.3 |
| 8764 | A>G
(Ala80Thr) | G | – | A | 26.7 |
| 8793 | T>C | T | C | C | |
| 8856 | G>A | G | A | A | |
| 8860 | A>G
(Thr112Ala) | A | G | G | 66.7 |
| COIII | 9540 | T>C | T | C | C | |
| ND3 | 10398 | A>G
(Thr114Ala) | A | G | G | 43.3 |
| 10400 | C>T | C | T | T | |
| ND4L | 10529 | A>G | A | – | G | |
| 10646 | G>A | G | A | A | |
| ND4 | 10873 | T>C | T | C | C | |
| 10203 | C>T | C | – | T | |
| 11674 | C>T | C | – | T | |
| 11719 | G>A | G | A | A | |
| 11778 | G>A
(Arg340His) | G | A | A | 98.3 |
| 12034 | C>T | C | T | – | |
| ND5 | 12549 | C>T | C | T | T | |
| 12705 | C>T | C | T | T | |
| 13135 | G>A
(Ala267Thr) | G | A | – | 5.0 |
| 13152 | A>G | A | G | G | |
| 13774 | A>G
(Thr480Ala) | A | – | G | |
| 14097 | C>T | C | T | – | |
| 14110 | T>C | T | – | C | |
| ND6 | 14502 | T>C
(Ile58Val) | T | C | C | 26.7 |
| CytB | 14766 | C>T
(Thr7Ile) | C | T | T | 50.0 |
| 14783 | T>C | T | C | C | |
| 15043 | G>A | G | A | – | |
| 15301 | G>A | G | A | A | |
| 15326 | A>G | A | G | G | |
| 15526 | C>T | C | – | T | |
Discussion
In the present study, according to the typical
clinical signs, symptoms and family histories, the probands were
diagnosed as having LHON (22,23).
Genetic and molecular characterization of the matrilineal relatives
was subsequently conducted. The G11778A mutations were observed in
all maternal members in the two families and were in the H2a2a1
haplogroup.
In previous reports, the average penetrance of
visual loss was 19.2% in 11 families, who were carrying only the
G11778A mutation, while the average penetrance of visual loss in
four families carrying the G11778A and T14502C mutations was 38.8%
(24,25–27).
In addition, the penetrances of visual loss in families carrying
T3394C and G11778A mutations have been reported as 38, 38, 44 and
56% (28). In the present study,
the penetrances of visual loss in family 1 and family 2 were 30.8
and 33.3%, respectively. This suggested that the G11778A mutation
itself was not sufficient to induce visual loss, while the second
mutations were. Subsequently, conservative analysis of the missense
mutations, which were identified in the genotype analysis of the
two families, was performed. The T3394C mutation was highly
conserved, while T14502C and the other missense variants were
potential polymorphisms. Therefore, the T3394C mutation may
cooperate with the G11778A variant to contribute to the penetrance
of LHON. Although the T14502C variant was identified as a potential
polymorphism, it has been previously reported to act
synergistically with the T11778C mutation (24).
The penetrances of visual loss were 30.8 and 33.3%
in the two families, which were lower than those carrying either
the T14502C or T3394C second mutation. It was suggested that the
H2a2a1 haplogroup had a protective effect on the phenotypic
manifestation of LHON, similar to a previous observation, in which
haplogroup H had a protective effect in families carrying the
G11778A mutation (13). However,
it is not possible to exclude the small sample size as a cause for
the deviation.
When comparing the probands of families 1 and 2,
age-at-onset was <20 years old, severe visual impairment was
present, and the G11778A mutation and H2a2a1 haplogroup were
identified in both families. In family 1, the proband carrying
T3394C and T14502C mutations, presented initially with loss of
vision in one eye. Loss occurred in the other eye shortly after,
indicating the rapid development of the disease in this patient.
However, in family 2 the proband only carried the T14502C mutation,
and vision loss began in his second eye 11 months following loss of
vision loss in his first eye. This indicates a slower development
of the disease compared with that in family 1. Therefore, we
hypothesized that carrying a second mutation increased the rate of
visual impairment. It was suggested that individuals with the two
second mutations in family 1 were more likely to have shorter
intervals between the loss of vision in the right and left eyes. It
is generally accepted that possessing more second mutations may
increase the penetrance of LHON (29), however, this was not observed in
the present study. Additional modifying factors including
environmental factors, lifestyle, estrogen levels and nuclear genes
may be important in LHON (30,31).
Cytoplasmic hybrid (cybrid) cell model experiments have previously
demonstrated that estrogen may reduce the production of reactive
oxygen species in complex I-defective LHON cybrids, improve cybrid
energetic competence and lead to coordinated activation of
mitochondrial biogenesis (32).
Additionally, estrogen receptor β has been observed to localize to
the human retinal ganglion cells and the retinal nerve fibre layer
(32). A previous study observed
that a female with Perrault syndrome, which leads to a reduction in
estrogen, also suffered a severe manifestation of LHON due to the
G11778A mtDNA mutation (33),
which further suggested that estrogen has an important function in
LHON. The variation in the PARL gene of LHON also suggested
that it may be involved in the penetrance of the disease (34).
In conclusion, in the small sample of patients
examined in the present study, patients with LHON who carry the
G11778A mutation were observed to carry two second mutations,
T14502C and T3394C, and were more likely to exhibit shorter
intervals between visual loss in the right and left eyes. In
addition, the H2a2a1 haplogroup may have a protective effect on the
phenotypic manifestation of LHON.
Acknowledgments
The authors would like to thank the patients and
family members who were involved in and Dr Changzheng Chen at
Renmin Hospital of Wuhan University (Wuhan, China) for his
cooperation.
References
|
1
|
Carelli V, Ross-Cisneros FN and Sadun AA:
Mitochondrial dysfunction as a cause of optic neuropathies. Prog
Retin Eye Res. 23:53–89. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fraser JA, Biousse V and Newman NJ: The
neuro-ophthalmology of mitochondrial disease. Surv Ophthalmol.
55:299–334. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sitarz KS, Chinnery PF and Yu-Wai-Man P:
Disorders of the optic nerve in mitochondrial cytopathies: new
ideas on pathogenesis and therapeutic targets. Curr Neurol Neurosci
Rep. 12:308–317. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Leber T: Ueber hereditaere und congenital
angelegte sehnervenleiden. Graefes Arch Clin Exp Ophthalmol.
17:249–91. 187
|
|
5
|
Wallace DC, Singh G, Lott MT, et al:
Mitochondrial DNA mutation associated with Leber’s hereditary optic
neuropathy. Science. 242:1427–1430. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mackey DA, Oostra RJ, Rosenberg T, et al:
Primary pathogenic mtDNA mutations in multigeneration pedigrees
with Leber hereditary optic neuropathy. Am J Hum Genet. 59:481–485.
1996.PubMed/NCBI
|
|
7
|
Brandon MC, Lott MT, Nguyen KC, et al:
MITOMAP: a human mitochondrial genome database-2004 update. Nucleic
Acids Res. 33:D611–D613. 2005. View Article : Google Scholar
|
|
8
|
Carelli V, La Morgia C, Valentino ML,
Barboni P, Ross-Cisneros FN and Sadun AA: Retinal ganglion cell
neurodegeneration in mitochondrial inherited disorders. Biochim
Biophys Acta. 1787:518–528. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jia X, Li S, Xiao X, Guo X and Zhang Q:
Molecular epidemiology of mtDNA mutations in 903 Chinese families
suspected with Leber hereditary optic neuropathy. J Hum Genet.
51:851–856. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yu-Wai-Man P, Griffiths PG and Chinnery
PF: Mitochondrial optic neuropathies-disease mechanisms and
therapeutic strategies. Prog Retin Eye Res. 30:81–114. 2011.
View Article : Google Scholar :
|
|
11
|
Brown MD and Wallace DC: Spectrum of
mitochondrial DNA mutations in Leber’s hereditary optic neuropathy.
Clin Neurosci. 2:134–145. 1994.
|
|
12
|
Tońska K, Kodroń A and Bartnik E:
Genotype-phenotype correlations in Leber hereditary optic
neuropathy. Biochim Biophys Acta. 1797:1119–1123. 2010. View Article : Google Scholar
|
|
13
|
Hudson G, Carelli V, Spruijt L, et al:
Clinical expression of Leber hereditary optic neuropathy is
affected by the mitochondrial DNA-haplogroup background. Am J Hum
Genet. 81:228–233. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Romero P, Fernández V, Slabaugh M, et al:
Pan-American mDNA haplogroups in Chilean patients with Leber’s
hereditary optic neuropathy. Mol Vis. 20:334–340. 2014.
|
|
15
|
Zhang AM, Jia X, Bi R, et al:
Mitochondrial DNA haplogroup background affects LHON, but not
suspected LHON, in Chinese patients. PloS One. 6:e277502011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Meng X, Zhu J, Gao M, et al: The analysis
of mitochondrial DNA haplogroups and variants for Leber’s
hereditary optic neuropathy in Chinese families carrying the
m.14484T >C mutation. Yi Chuan. 36:336–345. 2014.In Chinese.
PubMed/NCBI
|
|
17
|
Rieder MJ, Taylor SL, Tobe VO and
Nickerson DA: Automating the identification of DNA variations using
quality-based fluorescence re-sequencing: analysis of the human
mitochondrial genome. Nucleic Acids Res. 26:967–973. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang S, Wang L, Hao Y, et al: T14484C and
T14502C in the mitochondrial ND6 gene are associated with Leber’s
hereditary optic neuropathy in a Chinese family. Mitochondrion.
8:205–210. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kong QP, Yao YG, Sun C, Bandelt HJ, Zhu CL
and Zhang YP: Phylogeny of east Asian mitochondrial DNA lineages
inferred from complete sequences. Am J Hum Genet. 73:671–676. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
van Oven M and Kayser M: Updated
comprehensive phylogenetic tree of global human mitochondrial DNA
variation. Hum Mutat. 30:E386–E394. 2009. View Article : Google Scholar
|
|
21
|
Lange C, Feltgen N, Junker B,
Schulze-Bonsel K and Bach M: Resolving the clinical acuity
categories ̔hand motion̓ and ̔counting fingers̓ using the Freiburg
Visual Acuity Test (FrACT). Graefes Arch Clin Exp Ophthalmol.
247:137–142. 2009. View Article : Google Scholar
|
|
22
|
Carelli V, Ross-Cisneros FN and Sadun AA:
Mitochondrial dysfunction as a cause of optic neuropathies. Prog
Retin Eye Res. 23:53–89. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fraser JA, Biousse V and Newman NJ: The
neuro-ophthalmology of mitochondrial disease. Surv Ophthalmol.
55:299–334. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang J, Zhou X, Zhou J, et al:
Mitochondrial ND6 T14502C variant may modulate the phenotypic
expression of LHON-associated G11778A mutation in four Chinese
families. Biochem Biophys Res Commun. 399:647–653. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Qu J, Li R, Tong Y, et al: Only male
matrilineal relatives with Leber’s hereditary optic neuropathy in a
large Chinese family carrying the mitochondrial DNA G11778A
mutation. Biochem Biophys Res Commun. 328:1139–1145. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Qian Y, Zhou X, Hu Y, et al: Clinical
evaluation and mitochondrial DNA sequence analysis in three Chinese
families with Leber’s hereditary optic neuropathy. Biochem Biophys
Res Commun. 332:614–621. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Qu J, Zhou X, Zhang J, et al: Extremely
low penetrance of Leber’s hereditary optic neuropathy in 8 Han
Chinese families carrying the ND4 G11778A mutation. Ophthalmology.
116:558–564. e32009. View Article : Google Scholar
|
|
28
|
Zhang M, Zhou X, Li C, et al:
Mitochondrial haplogroup M9a specific variant ND1 T3394C may have a
modifying role in the phenotypic expression of the LHON-associated
ND4 G11778A mutation. Mol Genet Metab. 101:192–199. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Torroni A, Petrozzi M, D’Urbano L, et al:
Haplotype and phylogenetic analyses suggest that one
European-specific mtDNA background plays a role in the expression
of Leber hereditary optic neuropathy by increasing the penetrance
of the primary mutations 11778 and 14484. Am J Hum Genet.
60:1107–1121. 1997.PubMed/NCBI
|
|
30
|
Giordano C, Montopoli M, Perli E, et al:
Oestrogens ameliorate mitochondrial dysfunction in Leber’s
hereditary optic neuropathy. Brain. 134:220–234. 2011. View Article : Google Scholar :
|
|
31
|
Yu-Wai-Man P, Griffiths PG, Hudson G and
Chinnery PF: Inherited mitochondrial optic neuropathies. J Med
Genet. 46:145–158. 2009. View Article : Google Scholar :
|
|
32
|
Giordano C, Montopoli M, Perli E, et al:
Oestrogens ameliorate mitochondrial dysfunction in Leber’s
hereditary optic neuropathy. Brain. 134:220–234. 2011. View Article : Google Scholar :
|
|
33
|
Badura-Stronka M, Wawrocka A, Zawieja K,
Silska S and Krawczyński MR: Severe manifestation of Leber’s
hereditary optic neuropathy due to 11778 G A mtDNA mutation in a
female with hypoestrogenism due to Perrault syndrome.
Mitochondrion. 13:831–834. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Istikharah R, Tun AW, Kaewsutthi S, et al:
Identification of the variants in PARL, the nuclear modifier gene,
responsible for the expression of LHON patients in Thailand. Exp
Eye Res. 116:55–57. 2013. View Article : Google Scholar : PubMed/NCBI
|