Introduction
Oxidative stress-induced endothelial dysfunction is
involved in ischemia/reperfusion injury, which may further cause
fatal diseases, including coronary atherosclerosis caused by
myocardial infarction and stroke (1,2). It
has been well-established that endothelial progenitor cells (EPCs)
have a crucial role in angiogenesis of endothelial cells (ECs).
They can adhere to the endothelium at sites of hypoxia/ischemia and
participate in the formation of novel vessels through
differentiating into ECs.
Hypoxia has been shown to have effects on multiple
cellular biological processes of EPCs. For instance, hypoxia is
able to induce the mobilization of EPCs from the bone marrow into
the peripheral blood (3).
Furthermore, hypoxia can induce endothelial cells to secret
macrophage migration inhibitory factor (MIF), which may have a
crucial role in the recruitment and migration of EPCs to hypoxic
tissues (4). Lee et al
(5) showed that hypoxia inhibited
senescence of EPCs from aged individuals. The phosphoinositide-3
kinase (PI3K)/Akt signaling pathway has been found to have an
essential role in the regulation of cellular survival,
proliferation and migration. Moreover, the activity of the PI3K/Akt
signaling pathway is closely associated with hypoxia-induced EPCs.
Dai et al (6) showed that
hypoxia was able to protect against serum withdrawal-induced EPC
apoptosis, at least in part via the activation of the PI3K/Akt
pathway.
Hypoxia-inducible factor-1α (HIF-1α) is a key
transcriptional factor, the expression of which can be markedly
induced under hypoxia (7).
Moreover, HIF-1α has been shown to promote the proliferation and
differentiation of EPCs. Jiang et al (8) reported that inhibition of HIF-1α
suppressed the differentiation of EPCs to ECs, as well as the
expression of genes which promote neovascularization. However, the
regulatory pattern of HIF-1α under hypoxia in EPCs remains to be
fully elucidated.
Apelin is an endogenous ligand for the G
protein-coupled receptor APJ. The apelin/APJ pathway has a role in
the regulation of diverse biological functions, including
cardiovascular function, fluid homeostasis and insulin secretion,
via activation of various tissue-specific signaling pathways
(9–11). Recently, Ye et al (12) showed that the downregulation of the
serum levels of apelin was associated with acute myocardial
infarction-induced mobilization of EPCs, suggesting that apelin may
be involved in the regulation of EPCs. Furthermore, apelin
deficiency impaired sprouting of EPCs in ischemia-reperfusion
injury (13). However, to the best
of our knowledge, whether apelin has a role in the regulation of
hypoxia-induced EPC proliferation as well as the underlying
molecular mechanism have not been investigated, yet.
The present study mainly aimed to investigate the
role of apelin/APJ signaling in hypoxia-induced EPC proliferation
and the underlying molecular mechanisms were also investigated.
Materials and methods
Cell culture
EPCs were purchased from Nlunbio (Changsha, China).
The EPCs were cultured in endothelial growth medium 2 (EGM-2)
supplemented with 20% fetal bovine serum (FBS) both purchased from
Life Technologies (Carlsbad, CA, USA) at 37°C in a humidified
incubator containing 5% CO2. For performing cell
proliferation analysis, LY294002 (Sigma Aldrich, St. Louis, MO,
USA) and 740Y-P (Phoenix Pharmaceuticals, Belmont, CA, USA) were
used to treat EPCs in different groups.
Hypoxia treatment of EPCs
Prior to hypoxia treatment, EPCs were cultured in
serum-free EGM-2 at 37°C under 5% CO2 for 12 h. In the
MTT assay (Sigma-Aldrich), EPCs were cultured in EGM-2 containing
20% FBS at 37°C under 1% O2, 94% N2 and 5%
CO2 for 6, 12, 24 and 48 h, respectively.
Cell proliferation assay
The MTT assay was performed to determine cell
proliferation. In brief, MTT (10 mg/ml) was added to the medium.
After incubation for 4 h, the reaction was terminated by removal of
the supernatant and addition of 100 μl dimethyl sulfoxide
(DMSO; Sigma-Aldrich) to dissolve the formazan product. After 30
min, the optical density (OD) of each well was measured at 570 nm
using a plate reader (ELx808; Bio-Tek Instruments, Winooski, WT,
USA).
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR) analysis
TRIzol reagent (Life Technologies) was used to
extract total RNA from EPCs, in accordance with the manufacturer's
instructions. Total RNA was reverse transcribed into cDNA by using
the PrimeScript RT reagent kit, according to the manufacturer's
instructions (Takara Biotechnology Co., Ltd., Dalian, China). In
brief, 5 μl total RNA was mixed with 0.15 μl of 100
mm dNTPs (with dTTP), 1 μl (50 units) reverse transcriptase,
1.5 μl of X10 reverse transcription buffer, 0.19 μl
RNase inhibitor (20 U/μl), and nuclease-free H2O
was added to obtain a final volume of 15 μl. Reverse
transcription was performed at 16°C for 30 min, followed by an
incubation step at 42°C for 30 min and enzyme inactivation at 85°C
for 5 min. The mRNA expres-sion levels were determined using SYBR
Premix Ex Taq II, in accordance with the manufacturer's
instructions (Takara Biotechnology Co., Ltd.). In brief, 0.2
μl cDNA solution, 10 μl PCR mix, 2 μl gene
specific primer and 7.8 μl nuclease-free H2O were
mixed to obtain a final reaction volume of 20 μl. The
reaction conditions for PCR were: Pre-degeneration at 95°C for 1
min followed by 40 cycles of 95°C for 15 sec, 60°C for 15 sec, and
a final extension at 72°C for 1 min. The specific primer pairs were
as follows: HIF-1α sense, 5′-GAA CGT CGA AAA GAA AAG TCTCG-3′ and
anti-sense, 5′-CCT TAT CAA GAT GCG AAC TCACA-3′; apelin sense,
5′-GTC TCC TCC ATA GAT TGG TCTGC-3′ and anti-sense, 5′-GGA ATC ATC
CAA ACT ACA GCCAG-3′; APJ sense, 5′-CTC TGG ACC GTG TTT CGGAG-3′
and anti-sense, 5′-GGT ACG TGT AGG TAG CCCACA-3′; GAPDH (internal
reference) sense, 5′-GGA GCG AGA TCC CTCCAA AAT-3′ and anti-sense,
5′-GGC TGT TGT CAT ACT TCT CATGG-3′. Primers were purchased from
Shanghai Shenggong Co., Ltd. (Shanghai, China). Independent
experiments were repeated three times. The relative expres-sion of
mRNA was analyzed using the 2−ΔΔCt method.
Transfection
Transfection was performed using Lipofectamine 2000
(Invitrogen Life Technologies, Carlsbad, CA, USA) in accordance
with the manufacturer's instructions. All plasmids and siRNA were
purchased from Nlunbio. Sequences or other information were not
provided due to commercial confidentiality. Lipofectamine 2000,
siRNA and plasmid were diluted with serum-free medium,
respectively. The diluted Lipofectamine 2000 was added into the
diluted siRNA or plasmid and incubated for 20 min at room
temperature, and then added into the EPC suspension. Subsequently,
EPCs were incubated at 37°C, 5% CO2 for 6 h. After that,
the medium was replaced by Dulbecco's modified Eagle's medium with
10% FBS, and cultured for 24 h prior to the following assays.
Western blot analysis
Cells were solubilized in cold
radio-immunoprecipitation assay lysis buffer (Sigma-Aldrich).
Proteins were separated by 12% SDS-PAGE (Sigma-Aldrich) and
transferred onto a polyvinylidene difluoride (PVDF) membrane (Life
Technologies), which was then incubated with Tris-buffered saline
with Tween 20 (TBST; Sigma-Aldrich) containing 5% skimmed milk at
4°C for overnight. After that, the PVDF membrane was incubated with
specific primary antibodies, rabbit polyclonal anti-Apelin (cat.
no. ab59469; 1:100) and rabbit polyclonal anti-APJ (cat. no.
ab84296; 1:100) and incubated at room temperature for 3 h. After
washing with TBST three times, the PVDF membrane was incubated with
goat anti-rabbit IgG (cat. no. ab175734; 1:5,000, secondary
antibodies at room temperature for 1 h. All antibodies were
purchased from Abcam (Cambridge, MA, USA). After washing with TBST
three times, an enhanced chemiluminescence kit (Pierce Chemical,
Rockford, IL, USA) was used to perform chemiluminescent detection.
The relative expression of protein was determined by Image-Pro plus
software 6.0 (Media Cybernetics, Inc., Rockville, MD, USA)
represented as the density ratio versus GAPDH. The film was
purchased from Kodak (Tokyo, Japan).
Statistical analysis
Values are expressed as the mean ± standard
deviation of three independent experiments. Statistical analysis of
differences between values was performed using one-way analysis of
variance with SPSS 17 software (SPSS, Inc., Chicago, IL, USA).
P<0.05 was considered to indicate a statistically significant
difference between values.
Results
Hypoxia promotes EPC proliferation
First, the proliferation of EPCs was determined
under hypoxia and normoxia by using an MTT assay. As shown in
Fig. 1, after culturing under
hypoxia for 6, 12, 24 and 48 h, the relative proliferation of EPCs
was notably upregulated compared with that of EPCs cultured in
normoxia. These findings indicated that hypoxia promoted EPC
proliferation.
Hypoxia induces upregulation of HIF-1α
and apelin/APJ signaling
The expression levels of HIF-1α were determined in
each group, and it was found that hypoxia induced upregulation of
HIF-1α mRNA and protein expression in EPCs (Fig. 2A and B). As apelin and APJ are
known to be regulated by HIF-1α, their expression levels in EPCs
cultured in hypoxia or normoxia were then determined. As shown in
Fig. 3A and B, the mRNA and
protein levels of apelin and APJ were significantly upregulated in
EPCs cultured in hypoxia. In addition, ELISA was performed to
examine the secretion of apelin. As demonstrated in Fig. 3C, the secretion levels of apelin in
hypoxia-treated EPCs were also upregulated compared to those in
EPCs cultured under normoxia. These findings suggested that HIF-1α
as well as its downstream apelin/APJ signaling were upregulated in
hypoxia-treated EPCs.
Apelin/APJ signaling has a role in
hypoxia-induced EPC proliferation
The detailed role of apelin/APJ signaling in
hypoxia-induced EPCs proliferation was investigated. Four groups
were established: The normoxia group as a control, a hypoxia group,
a normoxia + apelin group and a hypoxia + apelin siRNA group. After
transfection of the EPCs with apelin plasmid or siRNA,
respectively, the expression levels of apelin were determined using
western blotting, and the results showed satisfactory transfection
efficiency (Fig. 4A). After that,
the role of APJ in the regulation of hypoxia-induced EPC
proliferation was investigated, for which four groups were
established: A normoxia group as the control, a hypoxia group, a
hormoxia + APJ group and a hypoxia + APJ siRNA group. After
transfection of EPCs with APJ plasmid or siRNA, respectively, the
expression levels of APJ were determined using western blotting,
and the results showed satisfactory transfection efficiency
(Fig. 4B). Subsequently, EPC
proliferation was determined in each group by using the MTT assay.
As shown in Fig. 4C, the
proliferation of EPCs in the normoxia + apelin group was higher
than that in the normoxia group, and the proliferation of EPCs in
the hypoxia + apelin siRNA group was lower than that in the hypoxia
group. Furthermore, as shown in Fig.
4D, the proliferation of EPCs in the normoxia + APJ group was
higher than that in the normoxia group, and the proliferation of
EPCs in the hypoxia + APJ siRNA group was lower than that in the
hypoxia group. These findings suggested that apelin/APJ signaling
has a role in hypoxia-induced EPC proliferation.
 | Figure 4(A) Western blot analysis was
performed to examine the protein expression of apelin in EPCs
transfected with apelin plasmid or apelin siRNA, respectively. (B)
Western blot analysis was performed to examine the protein
expression of APJ in EPCs transfected with APJ plasmid or APJ
siRNA, respectively. GAPDH was used as an internal reference.
Control cells were not transfected. **P<0.01 vs.
control. (C) An MTT assay was performed to determine the cell
proliferation of EPCs in each group at 6, 12, 24 and 48 h. Groups:
Normoxia, EPCs cultured under normoxia; Hypoxia, EPCs cultured
under hypoxia; Normoxia + Apelin, EPCs transfected with apelin
plasmid and cultured under normoxia; Hypoxia + Apelin siRNA, EPCs
transfected with apelin siRNA and cultured under hypoxia.
**P<0.01, vs. normoxia. (D) An MTT assay was
performed to determine the cell proliferation of EPCs in each group
at 6, 12, 24 and 48 h. Groups: Normoxia, EPCs cultured under
normoxia; Hypoxia, EPCs cultured under hypoxia; Normoxia + APJ,
EPCs transfected with APJ plasmid and cultured under normoxia;
Hypoxia + APJ siRNA, EPCs transfected with APJ siRNA and cultured
under hypoxia. **P<0.01, vs. normoxia. Values are
expressed as the mean ± standard deviation. EPC, endothelial
progenitor cell; siRNA, small interfering RNA; OD, optical
density. |
PI3K/Akt pathway is involved in
apelin/APJ-mediated hypoxia-induced EPC proliferation
The present study further determined the underlying
molecular mechanism by which apelin/APJ mediated EPC proliferation
under hypoxia. To determine whether the PI3K/Akt pathway was
involved in the apelin/APJ-mediated EPC proliferation, PI3K
inhibitor LY294002 and PI3K agonist 740Y-P were used. Four groups
were set as follows: The hypoxia group, the hypoxia + LY294002
group, the hypoxia + apelin siRNA group and the hypoxia + apelin
siRNA + 740Y-P group. As shown in Fig.
5, the proliferation of EPCs in the hypoxia + LY294002 group
was lower than that in the hypoxia group, suggesting that
inhibition of PI3K/Akt signaling suppressed hypoxia-induced EPC
proliferation. In addition, the proliferation of EPCs in the
hypoxia + apelin siRNA + 740Y-P group was higher than that in the
hypoxia + apelin siRNA group, suggesting that upregulation of
PI3K/Akt signaling reversed the inhibitory effect of apelin siRNA
on hypoxia-induced EPCs proliferation. These results suggested that
the PI3K/Akt pathway acts as a downstream effector of apelin/APJ
signaling in EPCs, and is involved in the apelin/APJ-mediated EPC
proliferation under hypoxia.
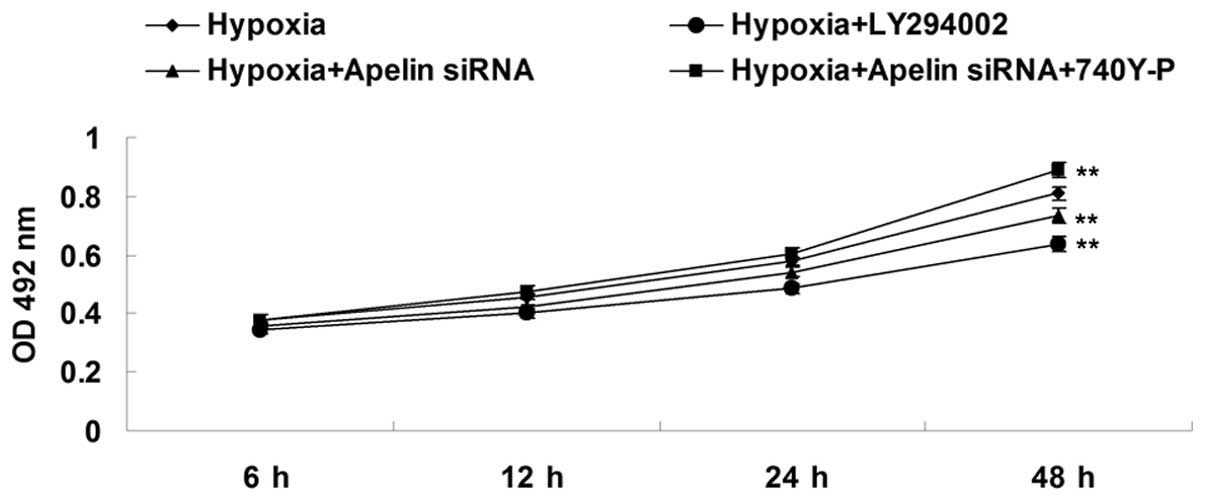 | Figure 5An MTT assay was performed to
determine the cell proliferation of EPCs in each group at 6, 12, 24
and 48 h. Groups: Hypoxia, EPCs cultured under hypoxia; Hypoxia +
Apelin siRNA, EPCs transfected with apelin siRNA and cultured under
hypoxia; Hypoxia + LY294002, EPCs treated with LY294002 and
cultured under hypoxia; Hypoxia + Apelin siRNA + 740Y-P, EPCs
transfected with apelin siRNA, treated with 740Y-P and cultured
under hypoxia. **P<0.01, vs. hypoxia. Values are
expressed as the mean ± standard deviation. OD, optical density;
siRNA, small interfering RNA; EPC, endothelial progenitor cell. |
Discussion
The present study showed that hypoxia induced
upregulation of HIF-1α and apelin/APJ signaling in EPCs. Increased
apelin/APJ further activated PI3K/Akt signaling, which was involved
in the upregulation of EPC proliferation. The findings of the
present study highlighted the importance of apelin/APJ signaling in
the regulation of EPC proliferation under hypoxia.
The critical role of EPCs in angiogenesis under
hypoxia/ischemia has been widely revealed. It has been well
established that hypoxia/ischemia triggers EPCs to migrate from the
bone marrow into the peripheral blood (14). After migration to the site of
hypoxic/ischemic tissues, EPCs can differentiate into ECs and
participate in the formation of novel vessels. HIF-1α is a key
determinant of oxygen-dependent gene regulation in angiogenesis,
which has been shown to be involved in EPC proliferation and
differentiation (8,15). For instance, several studies found
that inhibition of HIF-1α was able to inhibit the expression of
vascular endothelial growth factor (VEGF), VEGF receptor 2,
endothelial nitric oxide synthase as well as NO production, and
suppress the differentiation of EPCs to ECs (8,15).
On the contrary, overexpression of HIF-1α was reported to promote
the differentiation of EPCs to ECs (16). The present study showed that under
hypoxia, the expression of HIF-1α was significantly upregulated in
EPCs, consistent with the results of other studies (4,5).
Furthermore, the present study found that the expression of apelin
was upregulated under hypoxia in EPCs. In fact, the association
between HIF-1α and apelin has been elucidated by previous studies.
Ronkainen et al (17)
showed that the upregulation of apelin under hypoxia was abolished
by the HIF-inhibitory PAS protein in cardiomyocytes, suggesting
that the expression of apelin was regulated by hypoxia in cardiac
myocytes via the HIF pathway. Furthermore, Glassford et al
(18) showed that HIF-1α regulated
hypoxia-induced expression of apelin in adipocytes. However, to the
best of our knowledge, the association between HIF-1α and apelin in
EPCs has never been reported. As the results of the present study
showed that hypoxia induced the upregulation of HIF-1α as well as
apelin, it was hypothesized that the upregulation of HIF-1α induced
by hypoxia may further promote the expression of apelin in
EPCs.
Apelin/APJ has been found to be involved in a wide
range of physiological and pathological functions in the
cardiovascular system (19). For
instance, apelin has a crucial role in the regulation of cell
proliferation in vascular smooth muscle cells (VSMCs) (19). Moreover, inhibition of apelin
expression markedly inhibited EC proliferation in pathological
retinal angiogenesis (20).
Furthermore, apelin/APJ was found to increase angiogenesis and
improve cardiac functional recovery post-myocardial infarction
(21). These findings suggested
that Apelin has a key role in the regulation of cell proliferation
in the cardiovascular system. Recently, apelin was reported to be
involved in the mobilization of EPCs after acute myocardial
infarction (12). However, whether
apelin mediates EPCs proliferation has never been investigated, to
the best of our knowledge. The present study showed that
siRNA-mediated inhibition of apelin/APJ signaling markedly
attenuated EPC proliferation induced by hypoxia, indicating that
apelin/APJ signaling acts as a downstream regulator in
hypoxia-induced EPC proliferation.
Furthermore, it was found that inhibition of
PI3K/Akt signaling suppressed hypoxia-induced EPC proliferation,
while activation of PI3K/Akt signaling reversed the inhibitory
effect of apelin/APJ signaling knockdown on hypoxia-induced EPC
proliferation. These results confirmed that PI3K/Akt signaling was
the downstream effector of apelin/APJ signaling in the regulation
of EPC proliferation induced by hypoxia. Kleinz and Baxter
(22) showed that the protective
effect of apelin on myocardial reperfusion injury was independent
of PI3K/Akt signaling. However, Tao et al (23) reported that apelin protected the
heart against ischemia-reperfusion injury through activation of
PI3K/Akt signaling, via inhibition of endoplasmic reticulum
stress-dependent apoptosis activation. Moreover, another study also
showed that apelin protected against ischemic cardiomyocyte
apoptosis via activation of the PI3K/Akt pathway (24). Furthermore, Liu et al
(25) showed that apelin was able
to promote VSMC proliferation through activation of PI3K/Akt
signaling. In fact, most studies demonstrated that PI3K/Akt
signaling acted as a downstream effector of apelin, consistent with
the findings of the present study.
In conclusion, the present study reported that
hypoxia enhanced the expression of HIF-1α, which led to the
upregulation of apelin/APJ signaling, and further activated the
downstream PI3K/Akt signaling, an important promoter of EPC
proliferation. Therefore, apelin/APJ may serve as a potential
target for the prevention of hypoxic/ischemic injury in the
cardiovascular system.
References
|
1
|
Ganguly R, Lytwyn MS and Pierce GN:
Differential effects of trans and polyunsaturated fatty acids on
ischemia/reperfusion injury and its associated cardiovascular
disease states. Curr Pharm Des. 2013. View Article : Google Scholar
|
|
2
|
Kalogeris T, Baines CP, Krenz M and
Korthuis RJ: Cell biology of ischemia/reperfusion injury. Int Rev
Cell Mol Biol. 298:229–317. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schröder K, Kohnen A, Aicher A, et al:
NADPH oxidase Nox2 is required for hypoxia-induced mobilization of
endothelial progenitor cells. Circ Res. 105:537–544. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Simons D, Grieb G, Hristov M, et al:
Hypoxia-induced endothelial secretion of macrophage migration
inhibitory factor and role in endothelial progenitor cell
recruitment. J Cell Mol Med. 15:668–678. 2011. View Article : Google Scholar
|
|
5
|
Lee SH, Lee JH, Yoo SY, Hur J, Kim HS and
Kwon SM: Hypoxia inhibits cellular senescence to restore the
therapeutic potential of old human endothelial progenitor cells via
the hypoxia-inducible factor-1alpha-TWIST-p21 axis. Arterioscler
Thromb Vasc Biol. 33:2407–2414. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dai T, Zheng H and Fu GS: Hypoxia confers
protection against apoptosis via the PI3K/Akt pathway in
endothelial progenitor cells. Acta Pharmacol Sin. 29:1425–1431.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhu G, Tang Y, Geng N, et al:
HIF-alpha/MIF and NF-kappaB/IL-6 axes contribute to the recruitment
of CD11b+Gr-1+ myeloid cells in hypoxic microenvironment of HNSCC.
Neoplasia. 16:168–179. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jiang M, Wang CQ, Wang BY and Huang DJ:
Inhibitory effect of siRNA targeting HIF-1alpha on differentiation
of peripheral blood endothelial progenitor cells. Ai Zheng.
24:1293–1300. 2005.In Chinese.
|
|
9
|
Hashimoto T, Kihara M, Imai N, et al:
Requirement of apelin-apelin receptor system for oxidative
stress-linked atherosclerosis. Am J Pathol. 171:1705–1712. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bertrand C, Pignalosa A, Wanecq E, et al:
Effects of dietary eicosapentaenoic acid (EPA) supplementation in
high-fat fed mice on lipid metabolism and apelin/APJ system in
skeletal muscle. PLoS One. 8:e788742013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
O'Carroll AM, Lolait SJ, Harris LE and
Pope GR: The apelin receptor APJ: journey from an orphan to a
multifaceted regulator of homeostasis. J Endocrinol. 219:R13–R35.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ye J, Ni P, Kang L and Xu B: Apelin and
vascular endothelial growth factor are associated with mobilization
of endothelial progenitor cells after acute myocardial infarction.
J Biomed Res. 26:400–409. 2012. View Article : Google Scholar
|
|
13
|
Wang W, McKinnie SM, Patel VB, et al: Loss
of Apelin exacerbates myocardial infarction adverse remodeling and
ischemia-reperfusion injury: therapeutic potential of synthetic
Apelin analogues. J Am Heart Assoc. 2:e0002492013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Machalińska A: The role of circulating
endothelial progenitor cells in the development of vascular retinal
diseases. Klin Oczna. 115:158–162. 2013.In Polish.
|
|
15
|
Jiang M, Wang B, Wang C, et al: Inhibition
of hypoxia-inducible factor-1α and endothelial progenitor cell
differentiation by adenoviral transfer of small interfering RNA in
vitro. J Vasc Res. 43:511–521. 2006. View Article : Google Scholar
|
|
16
|
Jiang M, Wang CQ, Wang BY, He B, Shao Q
and Huang DJ: Overexpression of hypoxia inducible factor-1α
(HIF-1α) promotes the differentiation of endothelial progenitor
cell ex vivo. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 14:565–570.
2006.In Chinese. PubMed/NCBI
|
|
17
|
Ronkainen VP, Ronkainen JJ, Hänninen SL,
et al: Hypoxia inducible factor regulates the cardiac expression
and secretion of apelin. FASEB J. 21:1821–1830. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Glassford AJ, Yue P, Sheikh AY, et al:
HIF-1 regulates hypoxia- and insulin-induced expression of apelin
in adipocytes. Am J Physiol Endocrinol Metab. 293:E1590–E1596.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lv D, Li H and Chen L: Apelin and APJ, a
novel critical factor and therapeutic target for atherosclerosis.
Acta Biochim Biophys Sin (Shanghai). 45:527–533. 2013. View Article : Google Scholar
|
|
20
|
Kasai A, Ishimaru Y, Higashino K, et al:
Inhibition of apelin expression switches endothelial cells from
proliferative to mature state in pathological retinal angiogenesis.
Angiogenesis. 16:723–734. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li L, Zeng H and Chen JX: Apelin-13
increases myocardial progenitor cells and improves repair
postmyocardial infarction. Am J Physiol Heart Circ Physiol.
303:H605–H618. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kleinz MJ and Baxter GF: Apelin reduces
myocardial reperfusion injury independently of PI3K/Akt and P70S6
kinase. Regul Pept. 146:271–277. 2008. View Article : Google Scholar
|
|
23
|
Tao J, Zhu W, Li Y, et al: Apelin-13
protects the heart against ischemia-reperfusion injury through
inhibition of ER-dependent apoptotic pathways in a time-dependent
fashion. Am J Physiol Heart Circ Physiol. 301:H1471–H1486. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang Z, Yu B and Tao GZ: Apelin protects
against cardiomyocyte apoptosis induced by glucose deprivation.
Chin Med J (Engl). 122:2360–2365. 2009.
|
|
25
|
Liu C, Su T, Li F, et al: PI3K/Akt
signaling transduction pathway is involved in rat vascular smooth
muscle cell proliferation induced by apelin-13. Acta Biochim
Biophys Sin (Shanghai). 42:396–402. 2010. View Article : Google Scholar
|

















