Introduction
Autophagy is a self-degradative process whereby
organelles and cytoplasm are engulfed, digested and recycled to
sustain cell growth (1,2). Autophagy generally supports cancer
cell survival in response to metabolic stress; however, unstinted
autophagy causes progressive cellular consumption and eventually
cell death (3). Autophagy always
interplays with apoptosis (4).
Several stimuli cause apoptosis and also autophagy in the same
cell.
The 'Warburg effect' of cancer cells is
predominantly caused by the increased expression of the M2 isoform
of pyruvate kinase (PKM2), a rate-limiting enzyme, which catalyzes
the conversion of phosphoenolpyruvate (PEP) into pyruvate during
glycolysis (5,6). Different from PKL, PKR and PKM1, PKM2
forms not only tetramers (with high affinity for PEP), but also
dimers (with low affinity for PEP) (7). The expression levels of PKM2 are
increased in diverse types of human cancer and it is hypothesized
that PKM2 may be used as a clinical marker of cancer (8,9).
Downregulation of PKM2 promotes apoptosis (10,11);
however, the role of PKM2 in autophagy remains to be elucidated.
The present study investigated the effect of knocking down PKM2 on
autophagy and apoptotic cell death in A549 cells.
Materials and methods
Reagents
SYBR Premix Ex Taq, PrimeScript™ RT reagent kit,
gDNA Eraser and TRIzol reagent were purchased from Takara Bio Inc.
(Otsu, Japan). The
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
reagent, 3-methyladenine (3-MA), pLKO.1 vector, puromycin and mouse
anti-human anti-LC3 antibody (1:500; cat. no. L7543) were purchased
from Sigma-Aldrich (St. Louis, MO, USA). Chlorquine (CQ) was
obtained from InvivoGen (San Diego, CA, USA), and Dulbecco's
modified Eagle's medium (DMEM) and fetal bovine serum (FBS) were
purchased from Gibco Life Technologies (Carlsbad, CA, USA).
Cellulose Nitrate membranes and Luminata™ Crescendo Western
Horseradish Peroxidase (HRP) Substrate were purchased from
Millipore (Billerica, MA, USA). The protease and phosphatase
inhibitors were obtained from Roche (Basel, Switzerland). The
rabbit anti-human anti-PKM2 (1:500; cat. no. D78A4), rabbit
anti-human anti-cleaved caspase 3 (1:500; cat.no. 5A1E), rabbit
anti-human anti-Bcl-2 (1:500; 50E3), rabbit anti-human
anti-Beclin-1 (1:1,000; cat. no. D40C5) and rabbit anti-human
anti-actin (1:1,000; cat. no. 4967) antibodies were purchased from
Cell Signaling Technology, Inc. (Danvers, MA, USA). HRP-conjugated
donkey anti-mouse and anti-rabbit immunoglobulin G secondary
antibodies were purchased from Jackson Immuno Research
Laboratories, Inc. (West Grove, PA, USA). Lipofectamine 2000 and
fluorescein isothiocyanate (FITC) Annexin V/Dead Cell Apoptosis kit
with FITC-Annexin V and propidium iodide (PI) was purchased from
Invitrogen Life Technologies (Carlsbad, CA, USA). Caspase-Glo 3/7
assay was purchased from Promega Corporation (Madison, WI, USA) and
a Bicinchoninic Acid (BCA) Protein Assay kit was purchased from
DingGuo (Beijing, China).
Establishment of A549 short hairpin
(sh)PKM2-1 and A549 shPKM2-2 cells
The A549 cells were grown in monolayers at 37°C in
5% CO2 and maintained in DMEM, containing 100 units/ml
penicillin and 100 μg/ml streptomycin (Sangon, Shanghai,
China) sulfate, supplemented with 10% FBS. Two different target
sequences for human PKM2 were identified using BLOCK iT™ RNAi
designer: shPKM2-1, 5′-GCTGTGGCTCTAGACACTAAA-3′ and shPKM2-2,
5′-GTTCGGAGGTTTGATGAAATC-3′. These sequences were cloned into the
pLKO.1 vector (Sigma-Aldrich). The plasmids, shPKM2-1 and shPKM2-2,
were confirmed by DNA sequencing by Sangon. Lentiviral particles
were produced by co-transfecting the shPKM2-1 or shPKM2-2 vector
with VSVG and D8.9 packaging plas-mids (Addgene, Cambridge, MA,
USA) into HEK293T cells (American Type Culture Collection,
Manassas, VA, USA) using Lipofectamine 2000, according to the
manufacturer's instructions. The culture medium, which contained
viruses, was collected 48 h post-transfection and used to infect
cells. The infected A549 cells were selected in culture medium,
containing 4 μg/ml puromycin (Sigma-Aldrich) for 2
weeks.
Immunoblotting
Whole-cell lysates were extracted using
radioimmunoprecipitation lysis buffer, containing 50 mM Tris-HCl
(pH 8.0), 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate,
0.1% SDS and 2 mM MgCl2, supplemented with protease and
phosphatase inhibitors, and the protein concentration was
quantified using a BCA Protein Assay kit and a Nanodrop 1000
(Thermo Fisher Scientific, Waltham, MA, USA). The protein samples
(10 μg) were separated by 12% SDS-PAGE (Bio-Rad, Hercules,
CA, USA) and then transferred onto cellulose nitrate membranes,
followed by incubation in 5% non-fat milk for 1 h at room
temperature. The membrane was incubated in the primary antibody at
4°C overnight and subsequently incubated in the HRP-conjugated
secondary antibody at room temperature for 1 h. The target proteins
were detected by using Luminata™ Crescendo Western HRP Substrate
and the membranes were exposed to film.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The total RNA was extracted from cells using TRIzol
reagent, according to the manufacturer's instructions. Reverse
transcription of 1 μg total RNA was performed with the
PrimeScript™ RT reagent kit with gDNA Eraser, according to the
manufacturer's instructions. RT-PCR was performed in an Eppendorf
Mastercycler® ep realplex, according to the
manufacturer's instructions. The primers were as follows: Forward:
5′-GCACAGAGCCTCGCCTT-3′ and reverse: 5′-CCTTGCACATGCCGGAG-3′ for
β-actin; forward: 5′-GTGCGAGCCTCAAGTCACTCCACA-3′ and reverse:
5′-TATAAGAAGCCTCCACGCTGCCCA-3′ for PKM2; forward:
5′-GGTGGACCTGGAGAAGCTG-3′ and reverse: 5′-GGCACCCACATAAATGCC-3′ for
PFKL; forward: 5′-GTGCGCATGGGTATCTACG-3′ and reverse:
5′-ACTTGCAGGATGCTGGAGAC-3′ for PFKP; forward:
5′-GGCTGCGGCTGCTAACT-3′ and reverse: 5′-CAGGGCAATGTCAGACAACT-3′ for
ALDOC; forward: 5′-AGGGGTGTTTTTCCGTAAGC-3′ and reverse:
5′-GTTTCAAGCCATTCCTGCAT-3′ for ACYP1; and forward:
5′-TTGGCTGGGTGAAGAATACC-3′ and reverse: 5′-CTAGGGCTTCCAACCTTGCT-3′
for ACYP2. β-actin was used as an internal control. The comparative
threshold cycle (2−ΔΔCT) method was used to quantify the
mRNA expression levels of these genes.
Pyruvate kinase activity assay
A pyruvate kinase activity assay was performed using
2 mg cell lysates in 50 mM Tris-HCl, 100 mM KCl, 5 mM
MgCl2, 1 mM ADP, 0.5 mM PEP, 0.2 mM ADH and 8 units
lactate dehydrogenase at 37°C for 30 min. The reduction in
absorbance at 340 nm, attributable to the oxidation of NADH, was
measured using a microplate reader (Stat Fax-2100; Awareness
Technology Inc., Palm City, FL, USA) as the pyruvate kinase
activity.
MTT assay
The cells were plated into a 96-well plate at a
density of 5×103 cells/well. The following day, the
cells were treated with or without 2 mM 3-MA for 48 h. Medium,
supplemented with 0.5 mg/ml MTT was added to the cells. Following
culturing for 4 h, the MTT was replaced with 100 μl/well
dimethyl sulfoxide. The absorbance at 490 nm was detected using a
microplate reader (Awareness Technology Inc.).
Apoptosis analysis
The cells were pretreated with or without 2 mM 3-MA
for 48 h and harvested followed by washing in cold
phosphate-buffered saline. The washed cells were centrifuged at
1,000 × g and resuspended in 1X Annexin-binding buffer at a final
cell density of 1×106 cells/ml. A total of 100 μl
of the cells in annexin-binding buffer was obtained and mixed with
5 μl FITC/Annexin V (Component A) and 1 μl of 100
μg/ml PI working solution. Following incubation at room
temperature for 15 min, 400 μl of 1X Annexin-binding buffer
was added and the samples were maintained on ice. Apoptotic
analyses were performed by flow cytometry (BD FACSVerse; BD
Biosciences, Bedford, MA, USA), measuring the fluorescence emission
at 530 nm and >575 nm. Live cells exhibited only a low level of
fluorescence, apoptotic cells exhibited green fluorescence and dead
cells exhibited red and green fluorescence.
Caspase 3/7 activity assay
The cells were seeded into white-walled 96-well
plates to a final cell density at 2×103 cells/well and
were pretreated with or without 2 mM 3-MA for 48 h. The activity
levels of caspases 3/7 were analyzed with the Caspase-Glo 3/7
assay, according to the manufacturer's instructions. Briefly, the
plates were equilibrated to room temperature.
Caspase-Glo® 3/7 reagent (100 μl) was added into
each well. Following incubation at room temperature for 30 min, the
luminescent signal was detected with the Fluoroskan Ascent™
Microplate Fluorometer FL (Thermo Fisher Scientific).
Statistical analysis
The data are expressed as the mean ± standard
deviation. Student's t-tests were performed to determine
statistical significance of differences between experimental
groups, using GraphPad Prism 5 (GraphPad Software, Inc., La Jolla,
CA, USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
Knockdown of PKM2 in A549 cells using two
shRNAs
The A549 cells were infected with lentivirus,
containing shRNAs against PKM2. The expression levels of PKM2 were
analyzed by RT-qPCR and immunoblotting. Each of the shRNAs
efficiently decreased the mRNA and protein expression levels of
PKM2 in the A549 cells (Fig.
1).
Knockdown of PKM2 decreases the activity
of pyruvate kinase and glycolysis in A549 cells
Pyruvate kinase activity was detected using an
LDH-coupled enzyme assay and the mRNA expression levels of
glycolysis-associated enzymes were analyzed by RT-qPCR in A549
cells, with or without PKM2 knockdown. Downregulation of PKM2
significantly decreased the activity of pyruvate kinase and
glycolysis (Fig. 2).
Knockdown of PKM2 induces autophagy
The quantification of cells with punctate LC3 and
the expression levels of LC3II and Beclin-1 were examined by
immunoblotting to assess autophagy in the A549 cells with or
without PKM2 knockdown. Downregulation of PKM2 resulted in more
cells with punctate LC3, which indicated that autophagy was
induced. The expression levels of LC3II and Beclin-1 were
upregulated in A549 cells with PKM2 knockdown and use of an
autophagy inhibitor, CQ, increased the expression levels of LC3II
and Beclin-1 (Fig. 3).
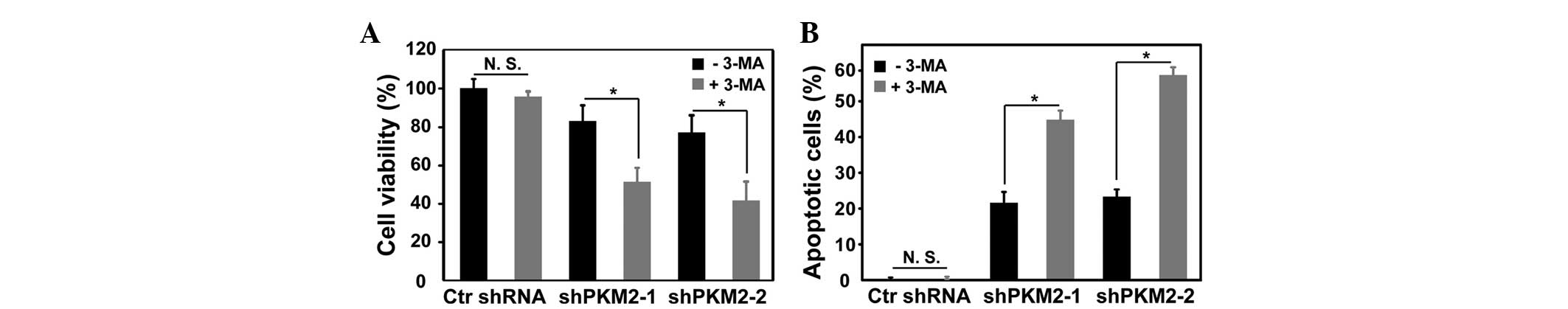
Inhibition of autophagy in A549 cells
with PKM2 knockdown promotes apoptotic cell death
Cell viability was detected using an MTT assay and
apoptosis was detected using Annexin V/PI staining and flow
cytometry in A549 cells with or without PKM2 knock down.
Downregulation of PKM2 induced apoptosis and autophagy in the A549
cells. Inhibition of PKM2 induced-autophagy increased apoptotic
cell death (Fig. 4).
Inhibition of autophagy in A549 cells
with PKM2 knockdown increases the activity of caspase 3/7 and
decreases the expression of Bcl-2
Inhibition of autophagy by 3-MA further increased
the activity of caspase 3/7 in A549 cells with PKM2 knockdown.
Downregulation of PKM2 increased caspase 3 cleavage and decreased
the expression of Bcl-2. Inhibition of autophagy using 3-MA caused
increased caspase 3 cleavage and markedly lower expression of Bcl-2
(Fig. 5).
Discussion
The metabolic hallmark of cancer cells, unlike
normal cells, is aerobic glycolysis (5,6).
Increased glucose uptake and increased glycolytic flux occur in
cancer cells. The increased conversion of glucose to lactate has
been detected in the majority of cancer cells, even under
oxygen-sufficient conditions, therefore, facilitating cell
proliferation (12). Understanding
the molecular mechanisms underlying aerobic glycolysis may assist
in controlling carcinogenesis by modulating the metabolic pathways
of cancer cells.
PKM2 can be used as a clinical marker of cancer,
since it is markedly expressed in cancer cells (8,9).
Unlike the other three PKs, PKM2 forms not only tetramers (with
high affinity for PEP), but also dimers (with low affinity for PEP)
(7). PKM2 is allosterically
activated by the glycolytic metabolite fructose-1,6-bisphosphate
(13,14). The release of PEP leads to the
formation of dimeric PKM2 and a reduction in its catalytic
activity, resulting in the accumulation of glycolytic metabolites.
These can be used for the synthesis of macromolecular building
blocks via the pentose phosphate pathway (15). PKM2 can switch between its dimeric
and tetrameric forms in cancer cells (7). If PKM2 forms dimers in cancer cells,
the marked expression of PKM2 shifts glucose metabolism to
macromolecule biosynthesis to meet the demand of accelerated cancer
cell proliferation. The knockdown of PKM2 impaired the mRNA
expression levels of glycolytic enzymes (Fig. 2B). AMPK is a master regulator for
energy-sensing pathways and is upstream of mTOR (16). The present study hypothesized that
the impaired glycolysis in A549 cells with PKM2 knockdown may
increase the ratio of AMP/ATP and activate AMPK, therefore,
inhibiting mTORC1 kinase activity and promoting autophagy.
Apoptosis and autophagy are pivotal cellular
processes, which are important for the maintenance of cell
homeostasis. There are several connections between apoptosis and
autophagy. One mechanism, which couples apoptosis and autophagy is
the interaction of Beclin-1 and Bcl-2 (17). Beclin-1, the mammalian orthologue
of yeast Atg6, promotes autophagosomal membrane nucleation
(18). The Bcl-2 family performs
anti-apoptotic roles in cells (19). Beclin-1 contains a BH3 domain,
which is necessary to bind the proteins of the Bcl-2 family
(20–22). The release of Bcl-2 from Beclin-1
activates autophagy (23). The
knockdown of PKM2 possibly affects the interaction between Beclin-1
and the Bcl-2 family, which may be caused by reduced expression of
Bcl-2. Understanding the intracellular mechanisms, which couple
apoptosis and autophagy under the control of PKM2 remain to be
elucidated.
In conclusion, to the best of our knowledge, the
present study provided the first evidence that downregulation of
PKM2 induced apoptosis and autophagy in A549 cells. Pharmacological
inhibition of autophagy promoted apoptotic cell death in A549 cells
with PKM2 knockdown. These results provided a novel strategy for
the treatment of cancer by simultaneously inhibiting PKM2 and
autophagy.
Acknowledgments
This study was supported by the China Postdoctoral
Science Foundation funded project (no. 124726) and Postdoctoral
Sponsorship in Henan Province (no. 2013036).
References
|
1
|
Levine B and Klionsky DJ: Development by
self-digestion: molecular mechanisms and biological functions of
autophagy. Dev Cell. 6:463–477. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mizushima N: Autophagy: process and
function. Genes Dev. 21:2861–2873. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Baehrecke EH: Autophagy: dual roles in
life and death? Nat Rev Mol Cell Biol. 6:505–510. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mariño G, Niso-Santano M, Baehrecke EH and
Kroemer G: Self-consumption: the interplay of autophagy and
apoptosis. Nat Rev Mol Cell Biol. 15:81–94. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Warburg O, Wind F and Negelein E: The
metabolism of tumors in the body. J Gen Physiol. 8:519–530. 1927.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Warburg O: On the origin of cancer cells.
Science. 123:309–314. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mazurek S: Pyruvate kinase type M2: a key
regulator of the metabolic budget system in tumor cells. Int J
Biochem Cell Biol. 43:969–980. 2011. View Article : Google Scholar
|
|
8
|
Bluemlein K, Gruning NM, Feichtinger RG,
Lehrach H, Kofler B and Ralser M: No evidence for a shift in
pyruvate kinase PKM1 to PKM2 expression during tumorigenesis.
Oncotarget. 2:393–400. 2011.PubMed/NCBI
|
|
9
|
Eigenbrodt E, Basenau D, Holthusen S,
Mazurek S and Fischer G: Quantification of tumor type M2 pyruvate
kinase (Tu M2-PK) in human carcinomas. Anticancer Res.
17:3153–3156. 1997.PubMed/NCBI
|
|
10
|
Goldberg MS and Sharp PA: Pyruvate kinase
M2-specific siRNA induces apoptosis and tumor regression. J Exp
Med. 209:217–224. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Spoden GA, Rostek U, Lechner S,
Mitterberger M, Mazurek S and Zwerschke W: Pyruvate kinase
isoenzyme M2 is a glycolytic sensor differentially regulating cell
proliferation, cell size and apoptotic cell death dependent on
glucose supply. Exp Cell Res. 315:2765–2774. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mendoza-Juez B, Martínez-González A, Calvo
GF and Pérez-García VM: A mathematical model for the
glucose-lactate metabolism of in vitro cancer cells. Bull Math
Biol. 74:1125–1142. 2012. View Article : Google Scholar
|
|
13
|
Christofk HR, Vander Heiden MG, Wu N,
Asara JM and Cantley LC: Pyruvate kinase M2 is a
phosphotyrosine-binding protein. Nature. 452:181–186. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dombrauckas JD, Santarsiero BD and Mesecar
AD: Structural basis for tumor pyruvate kinase M2 allosteric
regulation and catalysis. Biochemistry. 44:9417–9429. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gruning NM, Rinnerthaler M, Bluemlein K,
et al: Pyruvate kinase triggers a metabolic feedback loop that
controls redox metabolism in respiring cells. Cell Metab.
14:415–427. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hardie DG: AMP-activated protein kinase: a
cellular energy sensor with a key role in metabolic disorders and
in cancer. Biochem Soc Trans. 39:1–13. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Maiuri MC, Criollo A and Kroemer G:
Crosstalk between apoptosis and autophagy within the Beclin 1
interactome. EMBO J. 29:515–516. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
He C and Levine B: The Beclin 1
interactome. Curr Opin Cell Biol. 22:140–149. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dietrich JB: Apoptosis and anti-apoptosis
genes in the Bcl-2 family. Arch Physiol Biochem. 105:125–135.
1997.In French. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Maiuri MC, Le Toumelin G, Criollo A, et
al: Functional and physical interaction between Bcl-X(L) and a
BH3-like domain in Beclin-1. EMBO J. 26:2527–2539. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kang R, Zeh HJ, Lotze MT and Tang D: The
Beclin 1 network regulates autophagy and apoptosis. Cell Death
Differ. 18:571–580. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sinha S, Colbert CL, Becker N, Wei Y and
Levine B: Molecular basis of the regulation of Beclin 1-dependent
autophagy by the gamma-herpesvirus 68 Bcl-2 homolog M11. Autophagy.
4:989–997. 2008. View Article : Google Scholar : PubMed/NCBI
|