Introduction
Steatocystoma multiplex (SM; OMIM184500) is an
uncommon disorder, which is characterized by numerous skin-colored
subcutaneous cysts that originate from the pilosebaceous duct.
Although a number of sporadic cases have been reported, SM is
considered to be an autosomal dominant disorder. Certain SM
pedigrees have been identified with mutations in the keratin 17
(KRT17) gene, which have also been reported in patients with
pachyonychia congenital type 2 (1). These mutations are highly conserved
at the beginning sequence of helix 1A in the KRT17 gene, at which
any substitution or deletion may lead to a distortion of the
α-helical structure at the beginning of the 1A domain (2). Mutations in this location are
considered to have a significant effect on the assembly and/or
integrity of the keratin cytoskeleton in cells (3). The tissues affected include the outer
root sheath of the hair follicle, sebaceous glands, nail beds and
other appendages (4), which
explain the phenotypes of nail dystrophy and steatocystoma
multiplex that are observed in patients with pachyonychia
congenital type 2 (3,5).
The present study investigated a Chinese
four-generation autosomal dominant SM pedigree in order to identify
the causative mutation by means of polymerase chain reaction (PCR)
analysis and DNA sequencing. In addition, a literature review of
KRT17 gene mutations in SM pedigrees was performed to investigate
the KRTl7 gene mutation and the genotype-phenotype correlation.
Patients and methods
Pedigree analysis
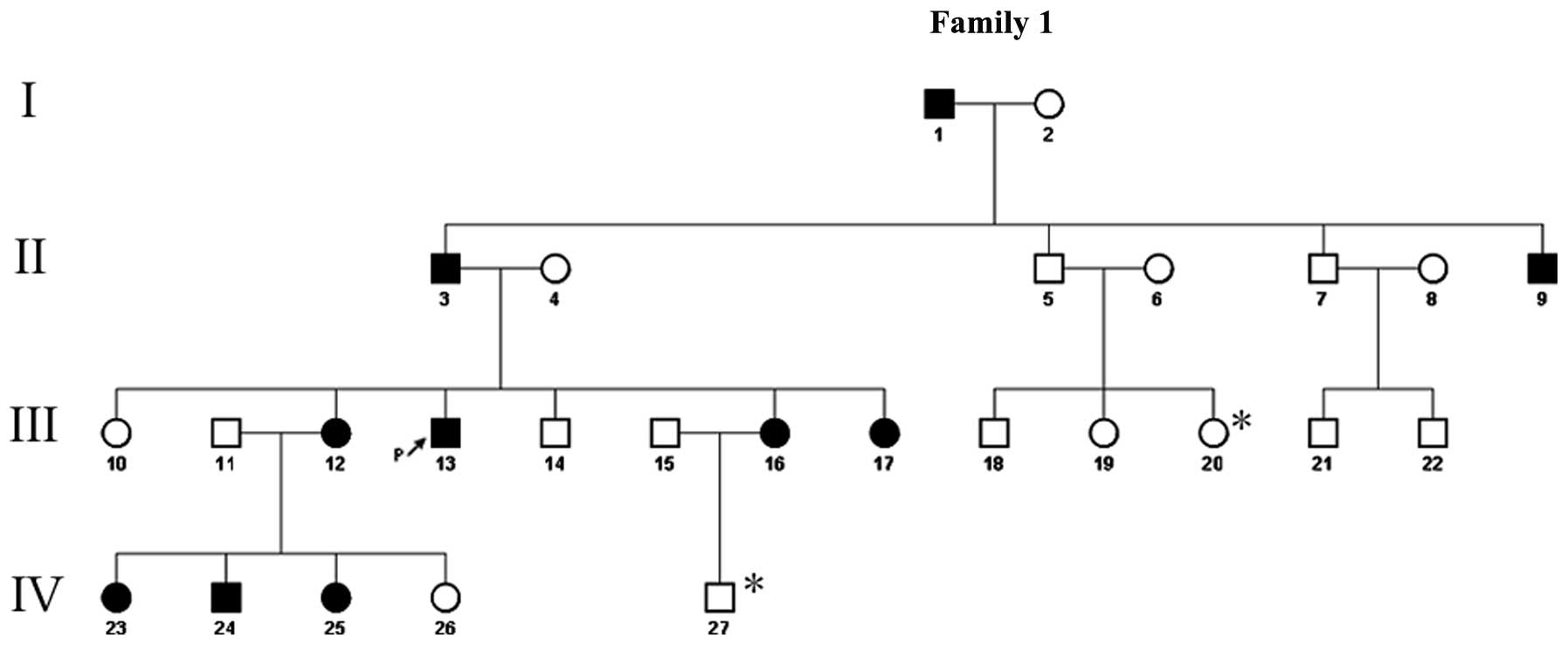
A four-generation Chinese SM pedigree with an
autosomal dominant mode of inheritance was examined. The
genealogical tree of this SM pedigree is shown in Fig. 1. The proband (III-13) was a
26-year-old male of Chinese Han-nationality, with the symptom of a
gradual appearance of multiple skin-colored nodules on his whole
body for >20 years. The lesions gradually developed in size and
number at infancy, and became severe and obvious during puberty.
The lesions were asymptomatic, however, were of cosmetic concern.
When punctured, the cysts discharged yellowish, oily material and
healed with a scar. The proband was in good general health and his
routine laboratory investigations were within normal limits. On
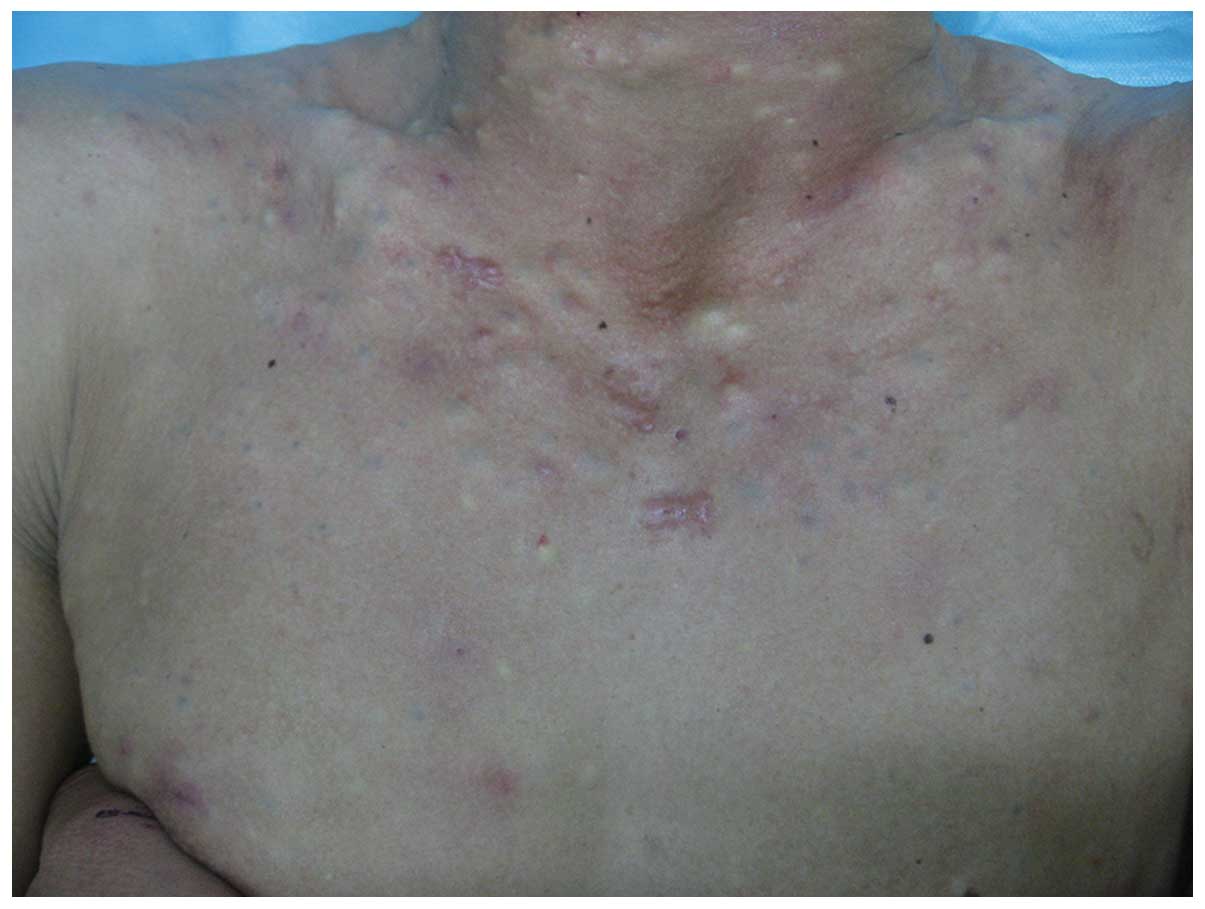
physical examination, multiple, skin-colored, firm, globoid
nodules, measuring between 2 and 30 mm in diameter on the face,
neck, chest, abdomen, arms and legs of the proband were identified
(Fig. 2). The nail, palmoplantar
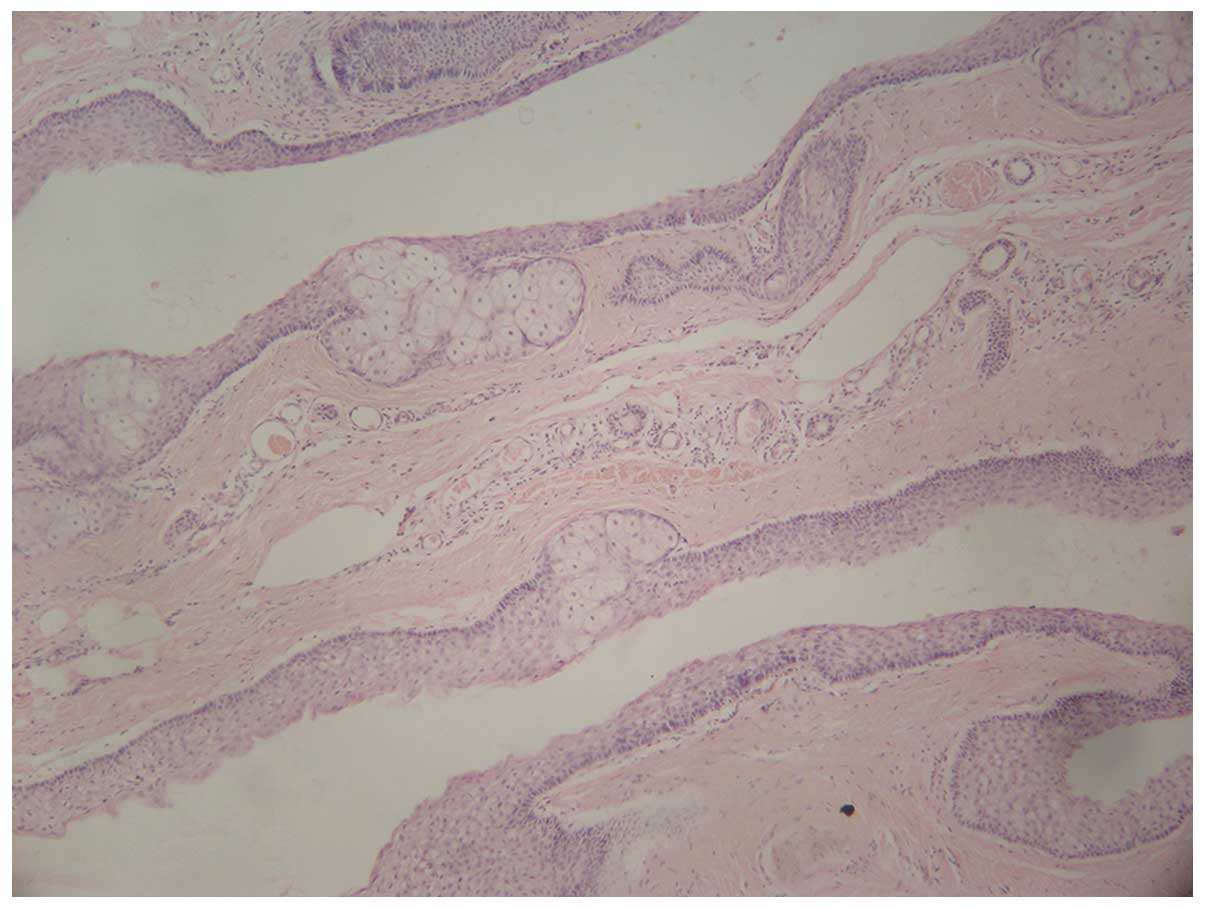
areas, mouth, tongue and teeth were all normal. Histological
examination of these lesions revealed that the cyst wall was
composed of several layers of epithelial cells, accompanied by
sebaceous gland lobules (Fig. 3).
On the basis of these findings, the diagnosis of SM was made.
Another nine affected members of his family reported a similar
history, namely, his grandfather (I-1), father (II-3), uncle
(II-9), three sisters (III-12,16,17), two nieces and one nephew
(IV-23,24,25). None of these affected individuals exhibited any
nail changes or any other skin, hair or mucosal abnormalities. The
conditions of the patients varied and the condition of the female
patients were comparatively milder in this family. The remaining
members of this family had no history of SM.
Mutation analysis
Ethical approval was obtained from the ethics
committee of Hainan Provincial Hospital of Skin Disease (Haikou,
China) and written informed consent was also obtained. The genomic
DNA was extracted from peripheral blood samples of 25 individuals
within the family (10 affected and 15 unaffected family members)
using a QIAamp blood-tissue kit (Qiagen, Hilden, Germany),
according to the manufacturer's instructions. The exon 1 coding
sequence of KRT17 (GenBank Entry: NM_000422) was amplified by
polymerase chain reaction (PCR) using primer pairs designed with
Primer3 (http://frodo.wi.mit.edu/primer3). The primer sequences
were as follows: Forward 5′-ATGGAAACAGAGGAGCA-3′ and reverse
5′-CCTGACTCAGCTTGCTGT-3′. The DNA template (100 ng) was mixed in a
solution containing 1X PCR buffer, consisting of 100 mM Tris-HCl
(pH 8.3) and 500 mM KCl, 1.5 mM MgCl2, 50 µM
dNTPs, 10 pmol of each primer, and 2.5 units of Taq DNA polymerase
(Promega Corporation, Madison, WI, USA), in a final reaction volume
of 30 µl. Amplification was performed in an initial
denaturation at 95ºC for 1 min, followed by 35 cycles of denaturing
at 95ºC for 40 sec, annealing at 58ºC for 40 sec and extension at
72ºC for 1 min, with a final extension at 72ºC for 3 min. This
resulted in a 830 bp fragment with the use of Eppendorf
Mastercycler Gradient PCR (Eppendorf, Hamburg, Germany). The PCR
samples contained 5% dimethyl sulfoxide to amplify the two
fragments. The PCR products were purified using a Promega
Wizard® SV 96 PCR Clean-up kit (Promega Corporation),
according to the manufacturer's instructions, eluted in 100
µl H2O and directly sequenced using an ABI Prism
377 DNA sequencer (Applied Biosystems Life Technologies, Foster
City, CA, USA). The mutation was confirmed by performing identical
PCR and sequencing in 100 unrelated control individuals. Geneious
version R8 software (Biomatters, Ltd., Auckland, New Zealand) was
used to compare sequences.
Literature review
A literature review of KRT17 gene mutations in SM
pedigrees was performed through searches of PubMed (http://www.ncbi.nlm.nih.gov/pubmed/), Ovid
Medline (http://ovidsp.tx.ovid.com/sp-3.16.0a/ovidweb.cgi),
EMBASE databases (http://www.embase.com) and China National Knowledge
Infrastructure (http://www.cnki.net/). To the best of
our knowledge, only seven types of KRT17 gene mutation in nine SM
pedigrees have been reported in the literature (2,3,5–17)
(Table I).
 | Table ISynopsis of KRT17 mutations in SM
pedigrees in Chinese non-Chinese populations from a review of
previous studies. |
Table I
Synopsis of KRT17 mutations in SM
pedigrees in Chinese non-Chinese populations from a review of
previous studies.
| Domain | Mutation | Phenotype | Heredity | Population | Reference |
|---|
| V1 | S24L 71C>T | SM | Familial | Chinese | (6) |
| 1A | N92H 274A>C | SM | Familial | | (7) |
| 1A | N92S 275A>G | SM | Familial | Chinese | (8) |
| | PC-2 | Familial | | (2) |
| | | Familial | | (3) |
| | | Familial | | (5) |
| | | Familial &
sporadic | | (7) |
| | | Familial | | (9) |
| | | Familial | | (10) |
| | | Familial | | (11) |
| | | Familial | | (12) |
| 1A | R94H 281 | SM | Familial | | (7) |
| G>A | SM | Familial | Chinese | (8) |
| | PC-2 | Sporadic | | (13) |
| 1A | R94C 280 | SM | Familial | Chinese | (14) |
| C>T | SM | Familial | | (15) |
| | PC-2 | Familial | | (3) |
| | | Familial | | (12) |
| 1A | R94G 280C>G | SM | Familial | Chinese | (16) |
| 2B | L371P 1112T>C | SM | Familial | | (17) |
Results
Identification of the large SM pedigree
with autosomal dominant inheritance
Starting from the aforementioned proband, a
four-generation pedigree was constructed through extensive
genealogical investigation (Fig.
1). A total of 10 (five male and five female) of the 27 family
members exhibited similar clinical manifestations. The availability
of the phenotypic data led to the establishment of a mode of
autosomal dominant inheritance in this large pedigree.
Identification of the causative gene
using PCR and DNA sequencing
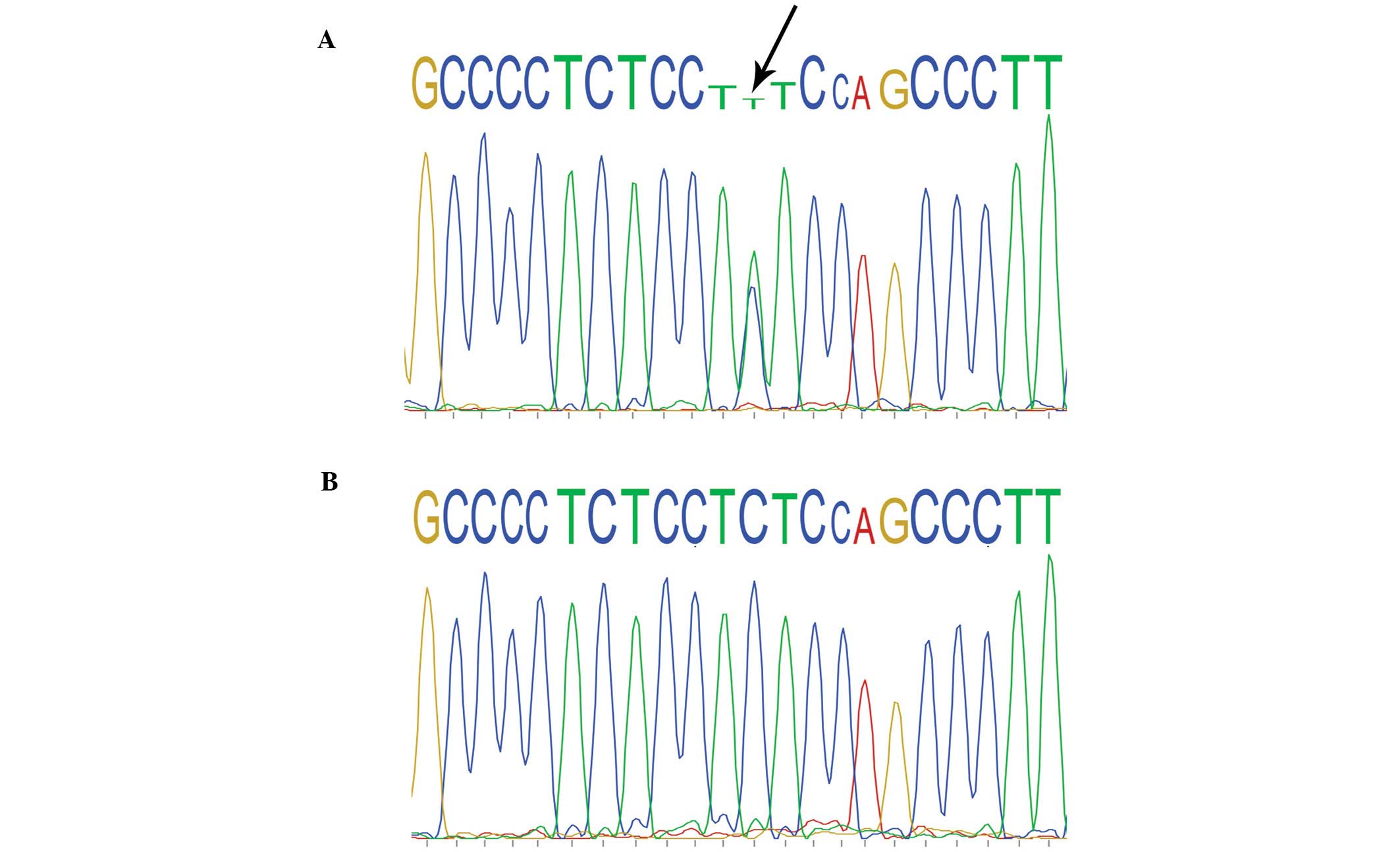
By direct sequencing of the KRT17 genomic PCR
products, a heterozygous mutation (c.280C>T) was identified in
exon 1 of the KRT17 gene in all 10 affected family members, which
causes an arginine to cysteine amino acid substitution at codon 94
(R94C). No such mutation was identified in any of the 15 unaffected
family members or in the 100 unrelated control individuals
(Fig. 4).
Discussion
The present study successfully identified the
heterozygous mutation, c.280C>T (R94C) in exon 1 of the KRT17
gene as the cause of SM in the large, four-generation Chinese
pedigree examined, and was revealed to be an autosomal dominant
form.
By reviewing the previous literature (2,3,5–17),
it was revealed that seven mutations have been previously reported
in nine families with SM (Table
I). These independent findings provided support for the
involvement of the KRT17 gene in SM. A total of three of seven
previously reported mutations, N92S, R94H and R94C, were associated
with SM and PC-2, while the other four, S24L, N92H, L371P and R94G,
were only associated with SM. S24L, N92S, R94H, R94C and R94G were
reported in seven Chinese families with SM, and the R94C mutation
has been detected previously in two families with SM and two
families with PC-2 (3,12,14,15),
which was the second time that SM was observed in a Chinese family.
The majority of mutations in familial SM have been reported within
the highly conserved helix boundary domains at the end of the
α-helical rod domain of keratin (7,8,14–16).
According to the data obtained in previous studies,
presented in Table I, a number of
different mutations may lead to the identical clinical phenotype in
SM, while the same mutations may cause different phenotypic
variations in patients with SM or PC-2. Previous studies indicated
variable onset ages of subcutaneous cysts in different SM pedigrees
or SM family members. For example, with the R94C mutation, the
onset of sebaceous cysts occurred in the same year as birth in
certain individuals, however, no sebaceous cysts occurred until the
second decade in other individuals (14,15).
This finding suggests that the genotype-phenotype correlation of SM
may be determined, not only by the site and type of the KRT17 gene
mutation, but also by other modifying factors. According to
previous studies, androgenic stimulation and environmental factors
have been suggested as the possible reasons (2,3,18).
In the pedigree examined in the present study, the early onset of
clinical manifestations, aggravation during puberty, variable
severity of different patients and milder symptoms in female family
members appeared to support the above-mentioned hypothesis that
there are other modifying factors contributing to the phenotype of
familial SM.
In conclusion, using PCR and DNA sequencing, the
present study identified a causative mutation, c.280C>T (R94C),
in the KRT17 gene in a large Chinese SM pedigree with an autosomal
dominant form. A review of the findings of previous studies
suggested that, with the exception of the mutation factor, other
modifying factors contribute to the phenotype of familial SM. This
observation may assist in elucidating the molecular consequences of
the KRT17 mutation in familial SM and may offer further insight
into the correlation between KRT17 gene mutations and
genotype-phenotype correlation.
Acknowledgments
The authors would like to thank the family members
recruited for their participation in the present study. The authors
would also like to thank Professor Yong Yang and Dr Zhimiao Lin
(Department of Dermatology, Peking University First Hospital,
Beijing, China) for assistance in DNA synthesis and sequencing.
References
|
1
|
McLean WH, Rugg EL, Lunny DP, Morley SM,
Lane EB, Swensson O, Dopping-Hepenstal PJ, Griffiths WA, Eady RA
and Higgins C: Keratin 16 and keratin 17 mutations cause
pachyonychia congenita. Nat Genet. 9:273–278. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Feng YG, Xiao SX, Ren XR, Wang WQ, Liu A
and Pan M: Keratin 17 mutation in pachyonychia congenita type 2
with early onset sebaceous cysts. Br J Dermatol. 148:452–455. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Covello SP, Smith FJ, Sillevis Smitt JH,
Paller AS, Munro CS, Jonkman MF, Uitto J and McLean WH: Keratin 17
mutations cause either steatocystoma multiplex or pachyonychia
congenita type 2. Br J Dermatol. 139:475–480. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Troyanovsky SM, Leube RE and Franke WW:
Characterization of the human gene encoding cytokeratin 17 and its
expression pattern. Eur J Cell Biol. 59:127–137. 1992.PubMed/NCBI
|
|
5
|
Smith FJ, Liao H, Cassidy AJ, Stewart A,
Hamill KJ, Wood P, Joval I, van Steensel MA, Björck E and
Callif-Daley F: The genetic basis of pachyonychia congenita. J
Investig Dermatol Symp Proc. 10:21–30. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ha WW, Wang J, Wang W, Fu HY, Tang HY,
Tang XF, Zhu J, Yin XY, Yang S and Zhang XJ: A novel missense
mutation of keratin 17 gene in a chinese family with steatocystoma
multiplex. Ann Dermatol. 25:508–510. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Smith FJ, Corden LD, Rugg EL, Ratnavel R,
Leigh IM, Moss C, Tidman MJ, Hohl D, Huber M and Kunkeler L:
Missense mutations in keratin 17 cause either pachyonychia
congenita type 2 or a phenotype resembling steatocystoma multiplex.
J Invest Dematol. 108:220–223. 1997. View Article : Google Scholar
|
|
8
|
Wang JF, Lu WS, Sun LD, Lv YM, Zhou FS,
Fang QY, Tang HY, Cui Y, Yang S and Zhang XJ: Novel missense
mutation of keratin in Chinese family with steatocystoma multiplex.
J Eur Acad Dermatol Venereol. 23:723–724. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fujimoto W, Nakanishi G, Hirakawa S,
Nakanishi T, Shimo T, Takigawa M and Arata J: Pachyonychia
congenita type 2: Keratin 17 mutation in a Japanese case. J Am Acad
Dermatol. 38:1007–1009. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cogulu O, Onay H, Aykut A, Wilson NJ,
Smith FJ, Dereli T and Ozkinay F: Pachyonychia congenita type 2,
N92S mutation of keratin 17 gene: Clinical features, mutation
analysis and pathological view. Eur J Pediatr. 168:1269–1272. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wilson NJ, Leachman SA, Hansen CD,
McMullan AC, Milstone LM, Schwartz ME, McLean WH, Hull PR and Smith
FJ: A large mutational study in pachyonychia congenita. J Invest
Dematol. 131:1018–1024. 2011. View Article : Google Scholar
|
|
12
|
Wilson NJ, Pérez ML, Vahlquist A, Schwartz
ME, Hansen CD, McLean WH and Smith FJ: Homozygous dominant missense
mutation in keratin 17 leads to alopecia in addition to severe
pachyonychia congenita. J Invest Dematol. 132:1921–1924. 2012.
View Article : Google Scholar
|
|
13
|
Terrinoni A, Smith FJ, Didona B, et al:
Novel and recurrent mutations in the genes encoding keratins K6a,
K16 and K17 in 13 cases of pachyonychia congenita. J Invest
Dematol. 117:1391–1396. 2001. View Article : Google Scholar
|
|
14
|
Wang X, Shi Y, Ye Y, Liu F, Jin W, Chen W,
Wang M, Hu L, Zhao G and Kong X: Keratin 17 gene mutation in
patients with steatocystoma multiplex. Zhonghua Yi Xue Za Zhi.
81:540–543. 2001.In Chinese.
|
|
15
|
Kamra HT, Gadgil PA, Ovhal AG and Narkhede
RR: Steatocystoma multiplex-a rare genetic disorder: A case report
and review of the literature. J Clin Diagn Res. 7:166–168.
2013.PubMed/NCBI
|
|
16
|
Zang D, Zhou C, He M, Ma X and Zhang J: A
novel mutation (p.Arg94Gly) of keratin 17 in a Chinese family with
steatocystoma multiplex. Eur J Dermatol. 21:142–144.
2011.PubMed/NCBI
|
|
17
|
Gass JK, Wilson NJ, Smith FJ, Lane EB,
McLean WH, Rytina E, Salvary I and Burrows NP: Steatocystoma
multiplex, oligodontia and partial persistent primary dentition
associated with a novel keratin 17 mutation. Br J Dermatol.
161:1396–1398. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Irvine AD and McLean WH: Human keratin
diseases: The increasing spectrum of disease and subtlety of the
phenotype-genotype correlation. Br J Dermatol. 140:815–828. 1999.
View Article : Google Scholar : PubMed/NCBI
|