Introduction
Liver sinusoidal endothelial cells (SECs) are highly
specialized fenestrated cells, without a basement membrane, which
constitute the walls of the liver sinusoid. They are unique among
other vascular endothelia (1).
They serve as the first contact of the liver with the hepatic blood
circulation, and the fenestrated structure is important for
filtering selected molecules and substances that enter the liver,
as well as controlling the exchange between the sinusoidal lumen
and the perisinusoidal space (space of Disse). Due to the fenestrae
and their lack of basement membrane, circulating lymphocytes come
into direct contact with hepatocytes (2).
Dysfunction of SECs is probably one of the initial
events in liver injury. Defenestration and capillarization of the
sinusoidal endothelium may be major contributors to hepatic failure
in cirrhosis (1). Studies have
shown close interactions between SECs and hepatic stellate cells
(HSCs), as SECs both prevent HSC activation and promote reversion
of activated HSCs to a non-activated phenotype (3). Therefore, preserving SEC fenestration
is essential for avoiding liver fibrosis and cirrhosis.
Endostar is a recombinant human endostatin
introduced by Chinese scientists (4,5).
Endostar was approved by the China State Food and Drug
Administration in 2005 for the treatment of non-small cell lung
cancer (4,6,7), and
is considered one of the most valuable anti-angiogenic agents. It
suppresses vascular endothelial growth factor (VEGF)-stimulated
proliferation, migration, and tube formation of human umbilical
vein endothelial cells in vitro (4). A study demonstrated that combined
treatment with Endostar and dexamethasone had synergistic effects
against experimental hepatoma growth (8). This combination may therefore provide
a novel strategy for improving the management of hepatoma or other
angiogenesis-dependent malignancies.
In a previous study, it was demonstrated that
Endostar decreased liver fibrosis and necrosis in a mouse model of
liver fibrosis induced by CCl4 and inhibited collagen
synthesis in the HSC-T6 rat stellate cell line in vitro
(9). The present study aimed to
further explore the antifibrogenic effects of Endostar by
investigating the impact of Endostar on SEC phenotype in
CCl4-induced fibrotic mice, and to better understand the
mechanisms underlying this action.
Materials and methods
Mouse model of CCl4-induced
liver fibrosis
The Animal Care and Use Committee of Harbin Medical
University (Harbin, China) approved all protocols and procedures.
Endostar was purchased from Simcere Pharmaceutical Research
(Nanjing, China). The animals were housed in an air-conditioned
room at 23-25°C with a 12 h dark/light cycle for one week prior to
initiation of the experiment. All animals received appropriate care
during the study with unlimited access to chow and water.
Male BALB/c mice weighing 18–20 g were obtained from
Beijing Vital River Laboratory Animal Technology (Beijing, China).
Liver fibrosis was induced in the remaining mice by intraperitoneal
injection (i.p.) of CCl4 (Beijing Brilliance Biochemical
Company, Beijing, China; 40% CCl4 in corn oil, 0.2
ml/100 g body weight, twice weekly) for 6 weeks as previously
reported (10). The mice were
divided into six treatment groups: Group 1, normal mice (n=7);
group 2, CCl4-induced liver fibrosis (n=10); group 3,
CCl4+Endostar (n=7; 20 mg/kg/day for 6 weeks, Endostar
was administered simultaneously with CCl4 injection for
6 weeks); group 4, CCl4+Endostar (n=7; 10 mg/kg/day for
6 weeks, Endostar was given simultaneously with CCl4
injection for 6 weeks); group 5, CCl4+Endostar (n=7; 20
mg/kg/day for 4 weeks, CCl4 only was administered to
mice for 2 weeks, then Endostar was given to mice simultaneously
with CCl4 injection for another 4 weeks); group 6,
CCl4+Endostar (n=7; 10 mg/kg/day for 4 weeks,
CCl4 only was administered to mice for 2 weeks, then
Endostar was given to mice simultaneously with CCl4
injection for another 4 weeks).
After the mice were euthanized (via 2% pentobarbital
i.p. at 0.3 ml/100 g body weight), liver samples were obtained from
all control mice and mice with CCl4-induced liver
fibrosis, with or without Endostar treatment. A section of the
liver was immediately snap-frozen in liquid nitrogen and stored at
−80°C until further use. Another section was embedded in paraffin
and sliced into 4 to 5-µm sections.
Transmission electron microscopy
(TEM)
Samples were processed for TEM as described
previously (11). Fresh specimens
were fixed in 3% glutaraldehyde, washed with phosphate-buffered
saline three times, and fixed in 1% osmic acid for 60 min. The
samples were dehydrated through an alcohol series, embedded in EPON
812 epoxy resin (Hede Biotechnology Co., Ltd., Beijing, China), and
then cut into 50-nm sections with an ultrathin microtome (EMUC7;
Leica Microsystems GmbH, Wetzlar, Germany). After staining with
uranyl acetate and lead citrate for 30 min, the sections were
observed under a transmission electron microscope (HITACHI H-7650,
Tokyo, Japan). Ten hepatic sinusoids with a diameter of 2-3
µm were randomly selected from each group and the average
number of fenestrae per hepatic sinusoid was determined.
Detection of protein levels of VEGF, KDR
and FLT1 by western blot analysis
Protein was extracted from liver samples (Protein
Extractor IV; DBI, Shanghai, China), homogenized, and assayed using
the bicinchoninic acid method (Pierce BCA Protein Assay kit; Thermo
Fisher Scientific, Rockford, IL, USA). Protein samples (40
µg) were resolved via SDS PAGE (80 V for 40 min on a 5%
acrylamide stacking gel (Beijing Biochemical Company) and 120 V for
70 min on a 10 or 15% running gel), and then transferred to a
nitrocellulose membrane (390 MA for 70 min or 80 V for 120 min;
Hybond-C Extra Membrane 45; Amersham Biosciences, Uppsala,
Sweden).
The membranes were soaked in Tris-buffered saline
(10 mM Tris-HCl and 250 M NaCl), that contained 5% non-fat powdered
milk and 0.1% Tween-20, for 2 h to block non-specific sites, and
incubated with primary antibody overnight at 4°C in blocking
solution. The antibodies were as follows: Rabbit polyclonal
anti-mouse VEGF (cat. no. sc-507), FLT1 (cat. no. sc-9029) and KDR
(cat. no. sc-504) diluted 1:200 with Tris-buffered saline with
Tween-20 (Santa Cruz Biotechnology, Santa Cruz, CA, USA); mouse
monoclonal anti-mouse ACTB (1:10,000; cat. no. SAB1403520;
Sigma-Aldrich, St. Louis, MO, USA) and horseradish peroxidase
(HRP)-linked goat anti-rabbit (cat. no. ZDR-5306) or anti-mouse
(cat. no. ZDR-5307) IgG (1:10,000; Zhongshan Goldbridge Biochemical
Company, Beijing, China). The resultant blots were washed and
incubated with secondary antibody (HRP-linked goat anti-rabbit IgG)
for 2 h at room temperature.
Immunoreactivity was visualized using an enhanced
chemiluminescence kit (Thermo Fisher Scientific). Films were
scanned using the Bio-Rad imaging system [ChemiDoc™ MP Imaging
System (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Sensitivity
comparison of the ChemiDoc™ MP Imaging System versus X-ray film was
performed using blots of serial dilution of transferrin].
Individual levels of the above protein expression were normalized
to β-actin.
Statistical analysis
The results are expressed as the mean ± standard
deviation. Statistical analyses were performed using analysis of
variance and the unpaired Student's t-test as appropriate.
P<0.05 was considered to indicate a statistically significant
difference. Statistical analyses were performed using SPSS version
17.0 software (SPSS Inc., Chicago, IL, USA).
Results
Ultrastructural changes in fibrotic mice
following Endostar treatment
There were significantly fewer fenestrae per SEC in
the fibrotic mice relative to those of the normal mice (P<0.01;
Table I; Fig. 1A and B). In all the groups
administered Endostar, the number of fenestrae was significantly
higher compared with the untreated fibrotic mice (P<0.01;
Fig. 1C–F). There were no
significant differences among the low dose and high dose of
Endostar groups.
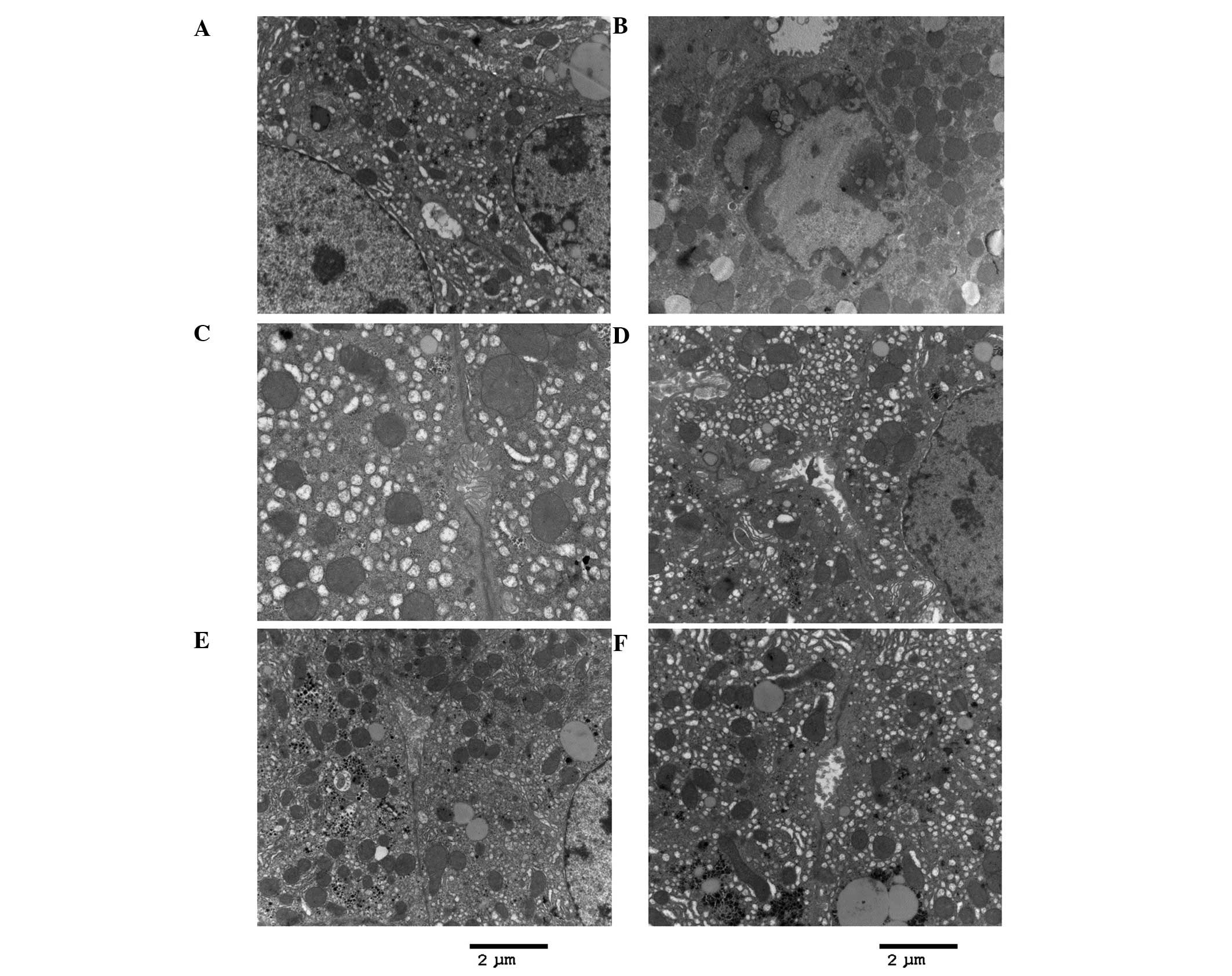
 | Figure 1CCl4-stimulated SEC
capillarization, basement and membrane formation, and decreased
hepatocyte microvilli. Endostar attenuated SEC capillarization and
increased hepatocyte microvilli. TEM slides of the liver from mice
in: (A) Group 1, normal mice (normal SECs and fenestrae); (B) group
2, CCl4-induced fibrosis group [fenestrae, cell
junctions (such as tight and ladder-like junctions on the cell
surface) and microvilli of hepatocytes reduced and a basement
membrane became apparent]; (C) group 3, Endostar 6-week group (high
dose): Fenestrae, microvilli and cell junctions between SECs were
similar to those of the normal control mice. TEM slides of the
liver from mice in: (D) Group 4, Endostar 6-week group (low dose):
Fenestrae, microvilli and cell junctions between SECs were similar
to those of the normal control mice. (E) TEM slide of the liver
from mice in the group 5, Endostar 4-week group (high dose):
Fenestrae were marginally decreased compared with those of the
normal control mice, the microvilli and cell junctions between SECs
became apparent. (F) TEM slide of the liver from mice in group 6,
Endostar 4-week group (low dose): Fenestrae and microvilli of
hepatocytes were marginally decreased compared with those of the
normal mice. (G and H) TEM slides of the liver from mice in group
2: A large quantity of collagen was generated. Stain, uranyl
acetate and lead citrate; scale bar, 2 µm. SEC, sinusoidal
endothelial cell; TEM, transmission electron microscopy. |
 | Table INumber of hepatic sinusoidal
endothelial cell fenestrae in the different groups. |
Table I
Number of hepatic sinusoidal
endothelial cell fenestrae in the different groups.
| Group (n) | Number of
fenestrae |
|---|
| Normal (neither
CCl4 nor Endostar) (7) | 7.43±0.98 |
| Model
(CCl4 alone) (8) | 2.38±0.91 |
| Lose-dose Endostar
(6 weeks) (5) | 4.60±0.90a |
| High-dose Endostar
(6 weeks) (5) | 4.80±0.84a |
| Low-dose Endostar
(4 weeks) (6) | 3.8±0.84a |
| High-dose Endostar
(4 weeks) (7) | 4.40±0.90a |
There were few or no collagenous fibers around the
hepatic central venules in the healthy control group. In the
untreated fibrotic model, mature collagen extended from the central
vein and the portal area to the hepatic lobules, and the basement
membrane was observed. In the Endostar groups, there were fewer
collagenous fibers compared with the model group (Fig. 1G and H).
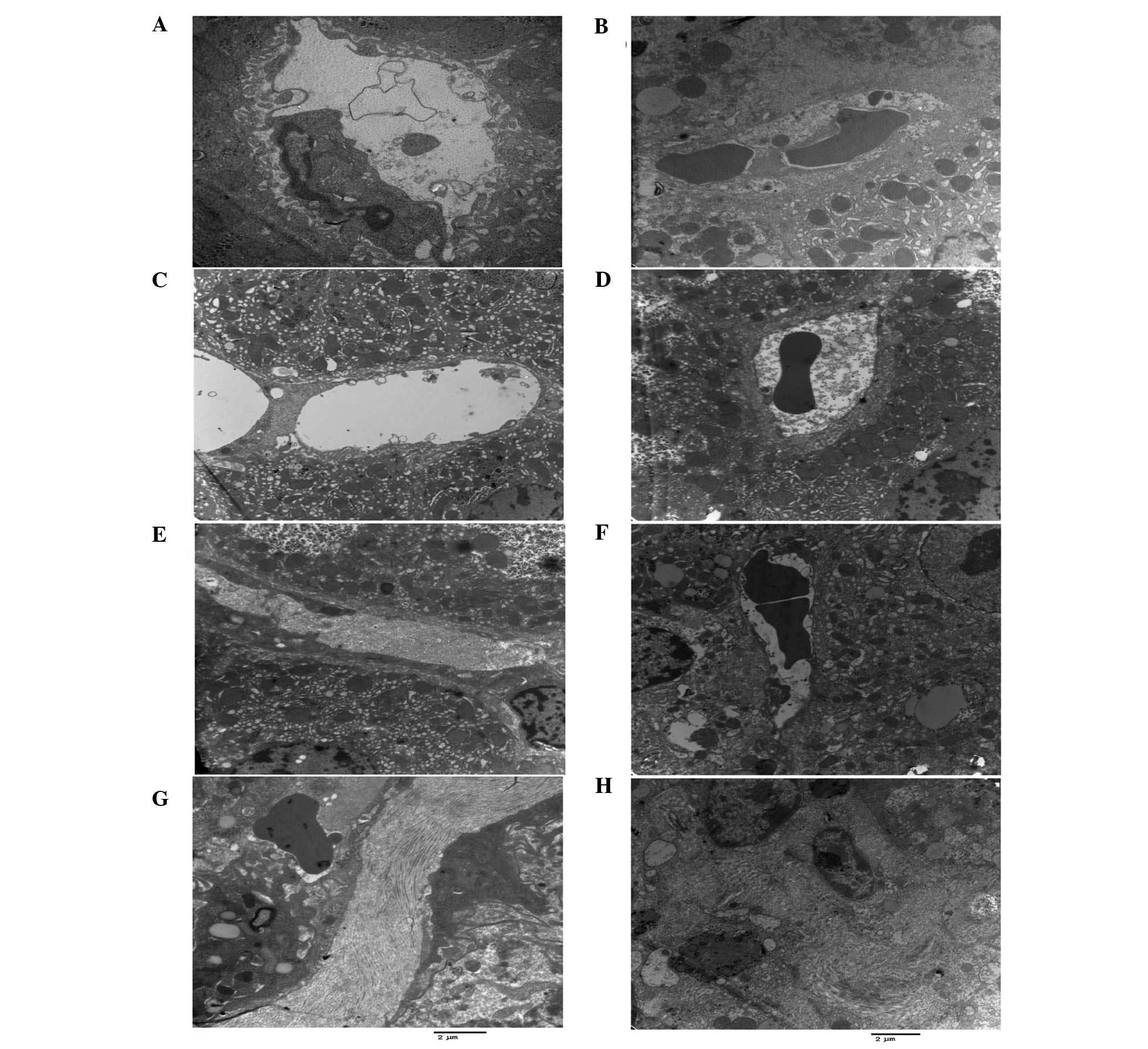
After 6 weeks of CCl4 exposure, the
junctions between hepatocytes in the untreated fibrotic models were
destroyed, and the microvilli of hepatocytes in the perisinusoidal
space and the intralobular bile ducts (cholangioles) disappeared
(Fig. 2B). By contrast, the
microvilli of hepatocytes were preserved in the Endostar groups
(Fig. 2C–F). Whereas expansion and
cholestasis were observed in the cholangioles of the fibrotic model
mice (CCl4 alone), they appeared normal in the Endostar
groups (Fig. 2). It was also
demonstrated that the extent of necrosis and inflammation in
hepatocytes was less in the Endostar groups compared with that in
the untreated model (Fig. 2).
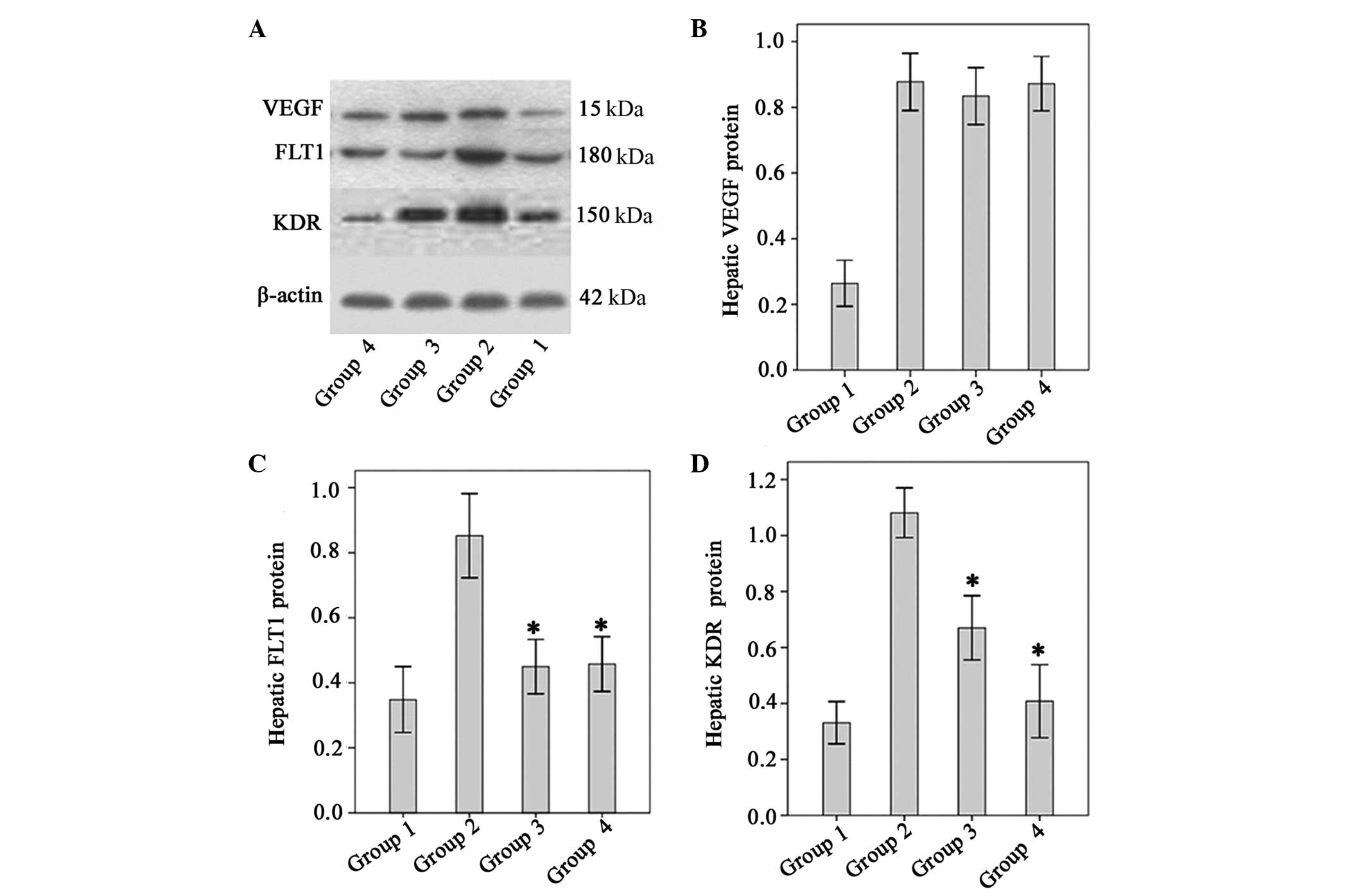
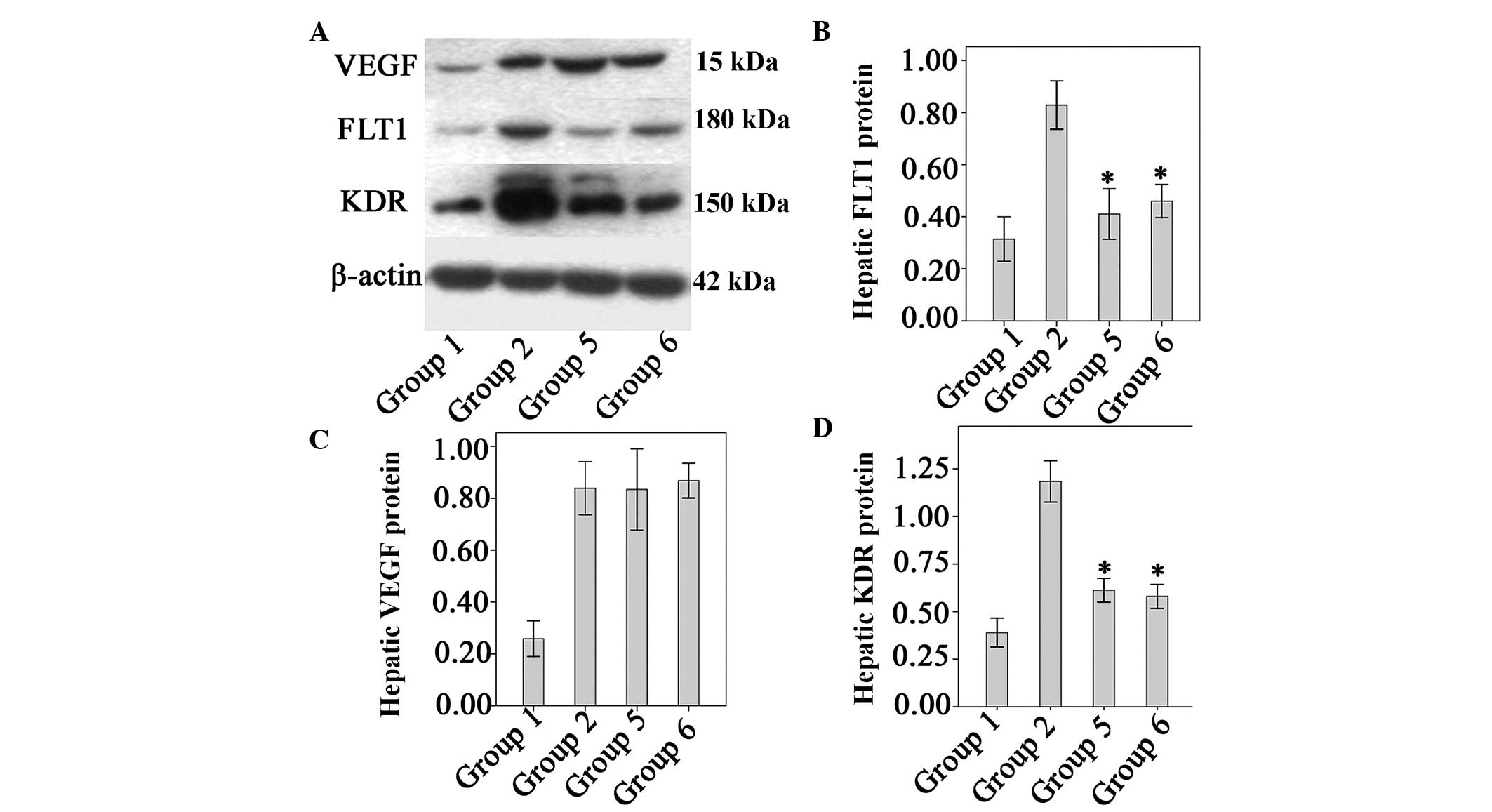
Effect of Endostar on VEGF, KDR and FLT1
protein levels
VEGF protein levels were significantly higher in the
fibrotic model mice than in the normal control mice (P<0.01).
There were no significant differences between the untreated
fibrotic mice and any of the groups treated with Endostar with
regard to VEGF expression (P>0.05, all; Fig. 4). The levels of FLT1 and KDR in
fibrotic model mice were significantly higher than those of the
normal controls. However, levels of FLT1 and KDR were significantly
lower in the four Endostar-treated groups relative to those of the
model (P<0.05; Fig. 5).
Discussion
Hepatic fibrosis is a major complication in chronic
liver diseases, increasing the risk of cirrhosis and ultimately
resulting in hepatic dysfunction and hepatocellular carcinoma.
Treating the cause of the liver disease may lead to fibrosis
reversal. However, resorting to anti-fibrotic compounds represents
an important complementary approach and a major therapeutic
challenge.
In the present study, it was demonstrated that
Endostar attenuates SEC capillarization and hepatic inflammation in
a mouse model of liver fibrosis induced by CCl4. In
addition, Endostar treatment was associated with reduced levels of
VEGFR protein in liver tissues, suggesting that Endostar may exert
its effects through the VEGF signaling pathway.
The results of the present study reinforce the
concept that angiogenesis exhibits a major role in liver
fibrogenesis. Evidence has been reported that angiogenesis
modulates the formation of liver fibrosis, as well as the
development of portal hypertension and hepatic carcinoma (12,13).
Intrahepatic angiogenesis and sinusoidal remodeling occur in a
number of chronic liver diseases. Anti-angiogenesis treatment may
be a therapeutic approach in portal hypertension (14). In addition, it has been reported
that anti-angiogenesis drugs, such as sorafenib and sunitinib,
which are used in the treatment of carcinoma, inhibit not only
hepatocellular carcinoma but also liver fibrosis (15–17).
Endostar is another anti-angiogenesis drug, which
targets the VEGF-induced tyrosine phosphorylation of VEGFR-2 and
ERK/MAPK signaling pathways in human umbilical vein endothelial
cells (4). Endostar has also been
shown to inhibit angiogenesis and hepatoma growth in vitro
(8). In the present study, it was
demonstrated that Endostar changed the SEC phenotype, leading to
attenuated sinusoidal capillarization. In parallel, microvilli
disappearance, inflammation, hepatocyte necrosis and bile duct
alterations were attenuated in all Endostar groups. These results
showed that anti-angiogenesis therapy may inhibit SEC sinusoidal
capillarization and attenuate hepatocyte damage.
VEGF promotes angiogenesis by binding to and
activating its receptors, FLT1 (VEGFR1), KDR (VEGFR2) and FLT4
(VEGFR3) (18). FLT1 and KDR are
predominantly expressed in endothelial cells and cancer cells,
while FLT4 is mainly expressed in lymphatic endothelial cells
(19–21). VEGR and VEGFRs (KDR and FLT1) are
important in the angiogenesis of the cirrhotic liver (22,23).
Upregulation of VEGF expression may be a stimulating factor of
angiogenesis in CCl4 and chronic bile duct ligation
induced-fibrosis models (24). In
the present study, increased VEGF, FLT1 and KDR expression after 6
weeks of CCl4 exposure was observed. Endostar was able
to inhibit FLT1 and KDR expression, but not VEGF expression.
Whether Endostar may be a VEGFR blocker and inhibit VEGF binding to
VEGFRs remains to be explored.
The molecular mechanism by which Endostar attenuates
liver injury remains unknown. SECs may be the primary target cell
in this process, and HSCs the secondary one. It has been well
documented that VEGF, FLT1, and KDR expression increases in rat
HSCs after CCl4 intoxication (25). Moreover, it is known that paracrine
signaling between SECs and HSCs modulates fibrogenesis,
angiogenesis and portal hypertension in chronic liver disease
(12,17,26,27).
It was also demonstrated that Endostar inhibited collagen synthesis
and downregulated tranforming growth factor-β1 expression in HSC-T6
cells in vitro (data not shown). The nature of the
interactions between SECs and HSCs in chronic liver disease after
Endostar treatment requires further investigation.
In conclusion, in the present study, Endostar
treatment was associated with hepatic sinusoidal endothelial cell
capillarization and reduced hepatocyte damage in
CCl4-induced fibrotic mice. These effects may involve
the VEGF pathway. Endostar is therefore a promising agent for
counteracting hepatic fibrosis. Further studies are required to
confirm its involvement in other causes of liver fibrosis and in
human chronic liver diseases.
Abbreviations:
|
SEC
|
sinusoidal endothelial cells
|
|
VEGF
|
vascular endothelial growth factor
|
|
VEGFR
|
vascular endothelial growth factor
receptor
|
|
TEM
|
transmission electron microscopy
|
Acknowledgments
This study was supported by the Postdoctoral
Foundation of Heilongjiang Province, China (grant no. LBH-Z11061),
Wang bao-en Liver Fibrosis Research Fund (grant no. 20100011) and
National Nature Science Foundation of China (grant no.
81170408).
References
|
1
|
Braet F and Wisse E: Structural and
functional aspects of liver sinusoidal endothelial cell fenestrae:
A review. Comp Hepatol. 1:12002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Iwakiri Y and Groszmann RJ: Vascular
endothelial dysfunction in cirrhosis. J Hepatol. 46:927–934. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Deleve LD, Wang X and Guo Y: Sinusoidal
endothelial cells prevent rat stellate cell activation and promote
reversion to quiescence. Hepatology. 48:920–930. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ling Y, Yang Y, Lu N, You QD, Wang S, Gao
Y, Chen Y and Guo QL: Endostar, a novel recombinant human
endostatin, exerts antiangiogenic effect via blocking VEGF-induced
tyrosine phosphorylation of KDR/Flk-1 of endothelial cells. Biochem
Biophys Res Commun. 361:79–84. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Whitworth A: Endostatin: are we waiting
for Godot? J Natl Cancer Inst. 98:731–733. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tong Y, Zhong K, Tian H, Gao X, Xu X, Yin
X and Yao W: Characterization of a monoPEG20000-Endostar. Int J
Biol Macromol. 46:331–336. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ni Q, Ji H, Zhao Z, Fan X and Xu C:
Endostar, a modified endostatin inhibits non small cell lung cancer
cell in vitro invasion through osteopontin-related mechanism. Eur J
Pharmacol. 614:1–6. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li XQ, Shang BY, Wang DC, Zhang SH, Wu SY
and Zhen YS: Endostar, a modifed recombinant human endostatin,
exhibits synergistic effects with dexamethasone on angiogenesis and
hepatoma growth. Cancer Lett. 301:212–220. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen J, Liu DG, Yang G, Kong LJ, Du YJ,
Wang HY, Li FD, Pei FH, Song JT, Fan YJ, et al: Endostar, a novel
human recombinant endostatin, attenuates liver fibrosis in
CCl4-induced mice. Exp Biol Med (Maywood). 239:998–1006. 2014.
View Article : Google Scholar
|
|
10
|
Wang H, Zhang Y, Wang T, You H and Jia J:
N-methyl-4-isoleucine cyclosporine attenuates CCl-induced liver
fibrosis in rats by interacting with cyclophilin B and D. J
Gastroenterol Hepatol. 26:558–567. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu H, Shi BM, Lu XF, Liang F, Jin X, Wu TH
and Xu J: Vascular endothelial growth factor attenuates hepatic
sinusoidal capillarization in thioacetamide-induced cirrhotic rats.
World J Gastroenterol. 14:2349–2357. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fernández M, Semela D, Bruix J, Colle I,
Pinzani M and Bosch J: Angiogenesis in liver disease. J Hepatol.
50:604–620. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Coulon S, Heindryckx F, Geerts A, Van
Steenkiste C, Colle I and Van Vlierberghe H: Angiogenesis in
chronic liver disease and its complications. Liver Int. 31:146–162.
2011. View Article : Google Scholar
|
|
14
|
Thabut D and Shah V: Intrahepatic
angiogenesis and sinusoidal remodeling in chronic liver disease:
New targets for the treatment of portal hypertension? J Hepatol.
53:976–980. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mejias M, Garcia-Pras E, Tiani C, Miquel
R, Bosch J and Fernandez M: Beneficial effects of sorafenib on
splanchnic, intrahepatic and portocollateral circulations in portal
hypertensive and cirrhotic rats. Hepatology. 49:1245–1256. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Majumder S, Piguet AC, Dufour JF and
Chatterjee S: Study of the cellular mechanism of Sunitinib mediated
inactivation of activated hepatic stellate cells and its
implications in angiogenesis. Eur J Pharmacol. 705:86–95. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Thabut D, Routray C, Lomberk G, Shergill
U, Glaser K, Huebert R, Patel L, Masyuk T, Blechacz B, Vercnocke A,
et al: Complementary vascular and matrix regulatory pathways
underlie the beneficial mechanism of action of sorafenib in liver
fibrosis. Hepatology. 54:573–585. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Olsson AK, Dimberg A, Kreuger J and
Claesson-Welsh L: VEGF receptor signalling-in control of vascular
function. Nat Rev Mol Cell Biol. 7:359–371. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Koch S and Claesson-Welsh L: Signal
transduction by vascular endothelial growth factor receptors. Cold
Spring Harb Perspect Med. 2:a0065022012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Koch S, Tugues S, Li X, Gualandi L and
Claesson-Welsh L: Signal transduction by vascular endothelial
growth factor receptors. Biochem J. 437:169–183. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Finley SD, Dhar M and Popel AS:
Compartment model predicts VEGF secretion and investigates the
effects of VEGF trap in tumor-bearing mice. Front Oncol. 3:1962013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yoshiji H, Kuriyama S, Yoshii J, Ikenaka
Y, Noguchi R, Hicklin DJ, Wu Y, Yanase K, Namisaki T, Yamazaki M,
Tsujinoue H, Imazu H, et al: Vascular endothelial growth factor and
receptor interaction is a prerequisite for murine hepatic
fibrogenesis. Gut. 52:1347–1354. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Moreau R: VEGF-induced angiogenesis drives
collateral circulation in portal hypertension. J Hepatol. 43:6–8.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Vanheule E, Geerts AM, Van Huysse J,
Schelfhout D, Praet M, Van Vlierberghe H, De Vos M and Colle I: An
intravital microscopic study of the hepatic microcirculation in
cirrhotic mice models: Relationship between fibrosis and
angiogenesis. Int J Exp Pathol. 89:419–432. 2008. View Article : Google Scholar
|
|
25
|
Ankoma-Sey V, Matli M, Chang KB, Lalazar
A, Donner DB, Wong L, Warren RS and Friedman SL: Coordinated
induction of VEGF receptors in mesenchymal cell types during rat
hepatic wound healing. Oncogene. 17:115–121. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Coulon S, Heindryckx F, Geerts A, Van
Steenkiste C, Colle I and Van Vlierberghe H: Angiogenesis in
chronic liver disease and its complications. Liver Int. 31:146–162.
2011. View Article : Google Scholar
|
|
27
|
Friedman SL: Preface. Hepatic fibrosis:
Pathogenesis, diagnosis and emerging therapies. Clin Liver Dis.
12:xiii–xiv. 2008. View Article : Google Scholar
|