Introduction
1q21.1 duplication syndrome is a rare aberration of
chromosome 1. Chromosome region 1q21.1 contains extensive and
complex low-copy repeats. Copy number variants (CNVs) in this
region are associated with developmental delay, neuropsychiatric
abnormalities, dysmorphic features and a variety of congenital
malformations (1,2). The characteristics of 1q21.1 deletion
[Online Mendelian Inheritance in Man (OMIM) no. 612474] include
developmental delay, microcephaly, facial anomalies, cataract and
congenital heart defect (CHD). Various anatomic types of CHD have
been found in 1q21.1 deletion syndromic patients (3), including left-sided obstructions as
well as conotruncal and septal defects (4–6). On
the other hand, 1q21.1 duplication (OMIM no. 612475) usually leads
to mental impairment, autism, macrocephaly and dysmorphic features
(1). In addition, a 1q21.1
deletion, as well as 1q21.1 duplication, have been found in
patients with apparently non-syndromic CHD. An association between
Tetralogy of Fallot (TOF) and 1q21.1 duplication or variants in
GJA5 gene mapping in 1q21.1 were documented recently
(7,8).
Latest developments of single nucleotide
polymorphism (SNP) array allow genome-wide screening at a
resolution (<5 Mb in size) that is undetectable by traditional
cytogenetic methods and have facilitated the discovery of a number
of novel microdeletion and microduplication syndromes (9). As part of a larger study on the
identification of pathogenic copy number variations in children
with CHD (10–12), a high-resolution Illumina SNP array
was performed on children with CHD and their parents. The present
study provided a detailed report on a Chinese female patient (age,
four years and eight months) with developmental delay,
neuropsychiatric abnormalities and CHD.
Materials and methods
Ethical approval and patient consent
The Review Board of the Second Xiangya Hospital of
the Central South University approved the present study (Changsha,
China). Written informed consent was obtained from the parents of
the patient for publication of the present study and any
accompanying images.
Clinical presentation
In 2011, a female patient (age, four years and eight
months) from Central-South China was seen at the Second Xiangya
Hospital of Central South University for a heart murmur. The
parents of the patient were unrelated and no family history of
inherited diseases was present. At birth, the patient presented
with lip cyanosis, clubbing and heart defects (tetralogy of fallot,
patent duct artery and patent foramen ovale). At the last
examination (at the age of four years and eight months), the
patient had a height of 75 cm and a weight of 10 kg. A moderate
mental impairment and a decreased ability were observed, and her
intelligence quotient was 65 compared to 80 at two years of age.
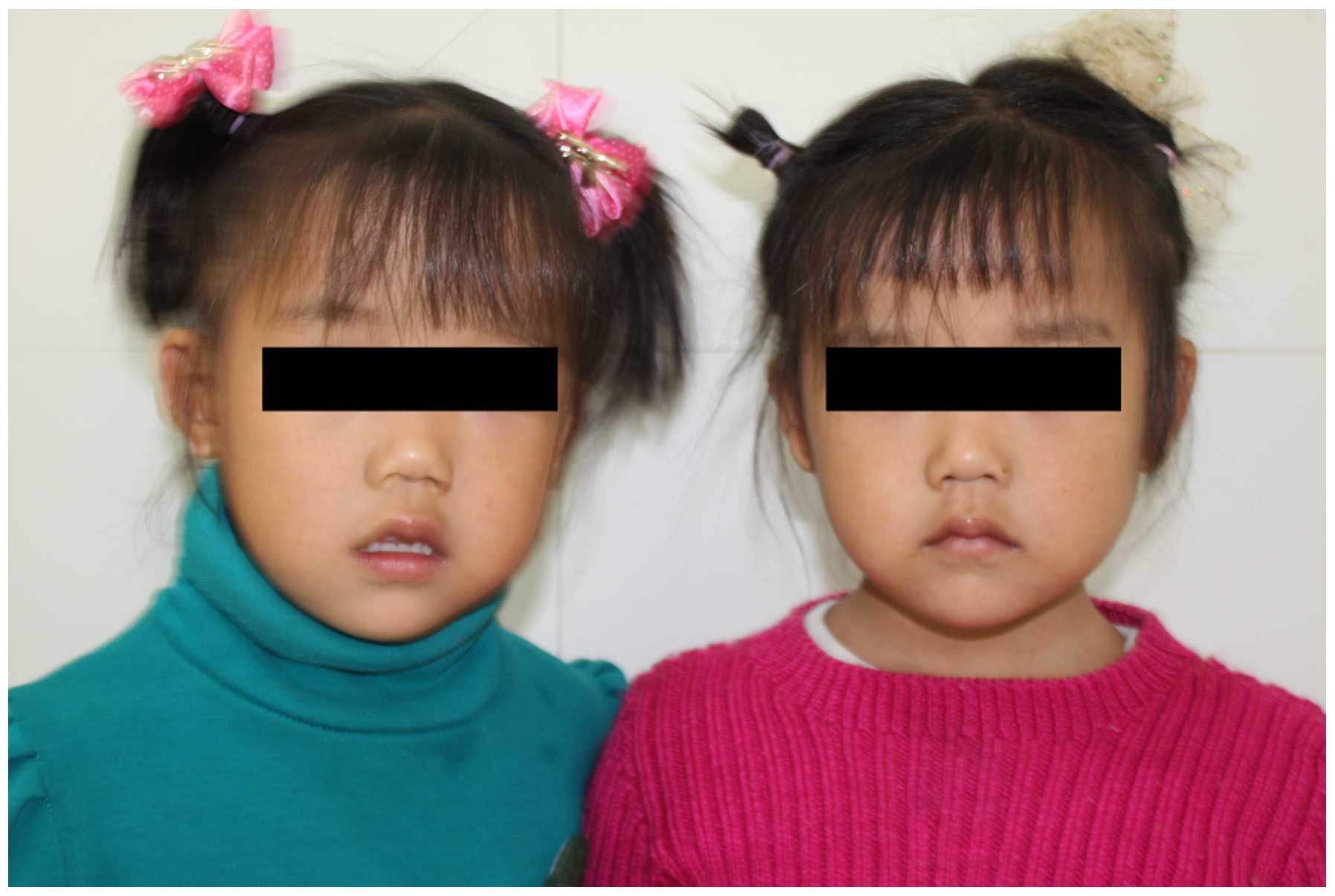
The patient did not present with any distinct facial abnormalities
(Fig. 1). The monozygotic twin
sister of the patient, however, was physically and psychologically
normal.
Cytogenetic analysis
Chromosome analysis was performed by conventional
G-banding techniques (550-band resolution) using the peripheral
blood samples of the patient, the twin sister and the parents. A
sample of 2 ml peripheral blood was collected. All samples were
subjected to lymphocyte culture according to a standard cytogenetic
protocol.
SNP array analysis
Peripheral venous blood was collected from the
patient and her family members. Genomic DNA was isolated from
peripheral blood leukocytes using a QIAamp DNA Blood Mini kit
(Qiagen, Valencia, CA, USA) according to manufacturer's
instructions and was adjusted to a final concentration of 100
ng/µl. The Human660w-Quad Chip (Illumina Inc., San Diego,
CA, USA) and the Illumina BeadScan genotyping system (Illumina
Inc.) were employed to obtain the signal intensities of probes
(SNP) following the manufacturer's instructions. The BeadStudio
3.3.7 software (Illumina Inc.) was used to analyze the genotypes
[human genome build 36.1(Hg18) for analysis] and evaluate the
experimental quality. The call rates (indicators of genotyping
fidelity) of the samples were >99.0%, indicating that the SNP
array results were reliable (11).
PCR-amplified short tandem repeat (STR)
analysis
STR analysis was performed on the Applied Biosystems
3130 Genetic Analyzer (Applied Biosystems Life Technologies, Foster
City, CA, USA) with the AmpFlSTR®
Identifiler® PCR Amplification kit (cat. no. 4322288;
Invitrogen Life Technologies, Carlsbad, CA, USA) according to the
manufacturer's instructions. Data were analyzed using Gene
Mapper® ID software (Life Technologies, Carlsbad, CA,
USA).
Results
The chromosome analysis of the patient revealed a
normal female karyotype, described as 46,XX. The twin sister and
parents also had a normal karyotype. STR-PCR analysis confirmed
that the patient and her sister are identical twins (Table I). Clinical examination of the
patient showed a combination of phenotypes with CHD, developmental
delay, mental impairment, neuropsychiatric abnormalities, raising
concerns about a chromosomal abnormality of
microdeletion/microduplication. To explore the exact genomic lesion
of this patient, the SNP array system (Human660w-Quad Chip,
Beadstation Scanner and BeadStudio 3.3.7 software) was employed to
analyze the whole genome copy number variations. Comparison with
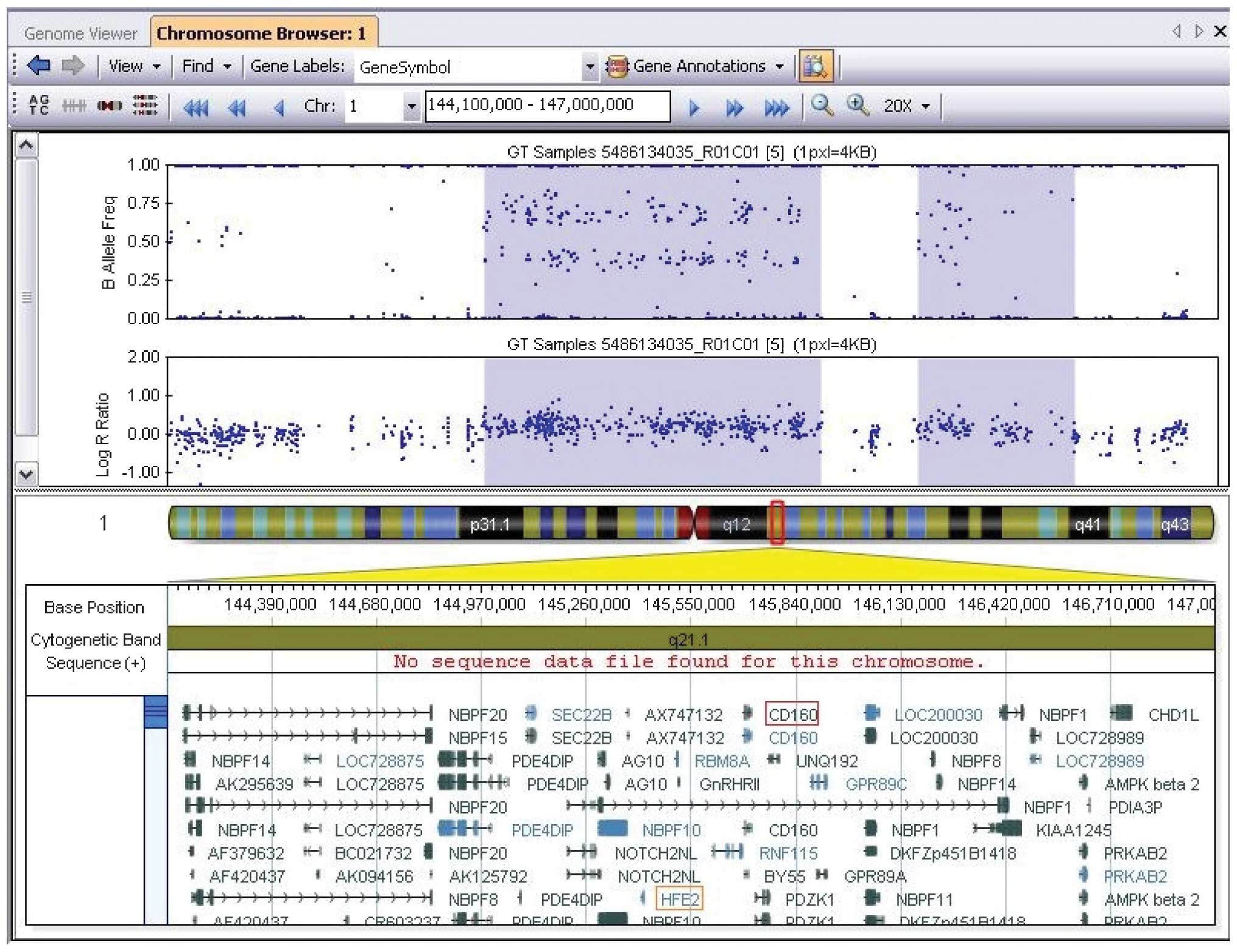
the Database of Genomic Variants (http://dgv.tcag.ca/dgv/app/home) showed a de
novo 1.6-Mb duplication at chromosome 1q21.1 (chromosome
1:144972830-146608260) (National Center of Biotechnology
Information build 37/Hg19). This chromosomal region contains ~30
annotated genes, including HFE2, HYDIN2, RBM8A
and CD160 (Fig. 2). The
identical twin sister and parents did not carry this genomic
lesion.
 | Table ISTR-polymerase chain reaction analysis
results. |
Table I
STR-polymerase chain reaction analysis
results.
| STR gene locus | Patient | Twin sister |
|---|
| D19S433 | 14.2, 17.2 | 14.2, 17.2 |
| D5S818 | 10, 11 | 10, 11 |
| D21S11 | 29, 32 | 29, 32 |
| D18S51 | 12, 14 | 12, 14 |
| D6S1043 | 10, 18 | 10, 18 |
| D3S1358 | 15, 16 | 15, 16 |
| D13S317 | 10, 10 | 10, 10 |
| D7S820 | 8, 11 | 8, 11 |
| D16S539 | 9, 12 | 9, 12 |
| CSF1PO | 11, 12 | 11, 12 |
| Penta D | 9, 14 | 9, 14 |
| vWA | 14, 14 | 14, 14 |
| D8S1179 | 12, 16 | 12, 16 |
| TPOX | 9, 10 | 9, 10 |
| Penta E | 11, 12 | 11, 12 |
| TH01 | 7, 9 | 7, 9 |
| D12S391 | 17, 22 | 17, 22 |
| D2S1338 | 23, 24 | 23, 24 |
| FGA | 23, 24 | 23, 24 |
Discussion
The present case report presented a Chinese female
patient (age, four years and eight months) with a de novo
microduplication of 1.6 Mb in 1q21.1. The patient had a normal
karyotype and presented with clinical phenotypes comprising several
malformations, developmental delay, neuropsychiatric abnormalities,
CHD (tetralogy of fallot, patent duct artery and patent foramen
ovale), a moderate mental impairment and a decreased ability. To
date, no evidence has suggested that children with cyanotic
congenital heart disease suffer from decreased mental performance;
therefore, the patient's mental impairment was unlikely to be due
to oxygen deprivation. However, the identical twin sister of the
patient who does not carry the same microduplication was physically
and psychologically normal. Using recent SNP array technology, the
patient was diagnosed with 1q21.1 duplication syndrome. To the best
of our knowledge, the present study was the first to report a
patient with 1q21.1 duplication syndrome in mainland China.
The chromosome 1q21.1 locus is a complex region with
multiple low-copy repeats that make the region susceptible to
recurrent deletions and duplications. Large rare copy number
variants (CNVs) at this locus as well as microdeletions and
microduplications have recently been associated with genomic
disorders (OMIM nos. 612474 and 612475), characterized by
developmental delay, neuropsychiatric abnormalities, dysmorphic
features and a variety of congenital malformations (1,2,13,14).
Two main classes of 1q21.1 CNVs, spanning 20–40 genes, have been
described. The more common one (class I; ~1.8 Mb) includes only the
distal 1q21.1 region, whereas the longer one (class II, ~2.7 Mb)
extends proximally to encompass the thrombocytopenia absent radius
(TAR) syndrome region (2).
Congenital heart defect is a major feature of 1q21.1 deletion
(3,5,6), and
has been occasionally reported in association with 1q21.1
duplication (8,15). The prevalence of CHD in a reported
series of 1q21.1 deletions was ~30% (3). The anatomic types were heterogeneous,
mainly comprising left-sided obstructions (40%), including aortic
coarctation, bicuspid aortic valve and subaortic stenosis, but also
septal defects (27%) and conotruncal anomalies (20%) (1). Of note, 1q21.1 duplication was more
common in patients with tetralogy of fallot (8).
In the 1q21.1 locus, the genes GJA5 (7), CHD1 L (8) and PRKAB2 (16) were reported to be closely
associated with CHD. The observation that none of them were
included in the base sequence of the proband inferred that there
may be other genes responsible for the occurrence of CHD. In the
present study, the identified chromosome region contained ~30
annotated genes, including HFE2, HYDIN2, RBM8A
and CD160. To date, only few studies have associated
duplications of these genes with CHD (17,18);
the findings of the present study led to the hypothesis that the
duplication of these genes caused an increased expression of the
coded proteins, which in turn led to CHD. HFE2, which is
also in this locus, encodes hemojuvelin (HJV), a protein involved
in the activation of hepcidin and iron metabolism. Primarily, HJV
is expressed in skeletal muscles, but it is also present at lower
levels in the heart and the liver (17). Furthermore, a soluble form of HJV
circulates in plasma. Mutations of this gene are known to be
associated with hereditary juvenile hemochromatosis (17,18).
The gene HYDIN2, a paralogous segment of the primary ciliary
dyskinesia-associated gene HYDIN (19), is involved in the development and
formation of organ barriers, and its abnormity also causes
congenital heart defects. Deletion/mutation of RBM8A can
cause TAR syndrome, characterized by a series of phenotypes
including coarctation of the aorta, left ventricular hypertrophy
and subendocardial fibrosis (20);
however, it has remained elusive whether duplication of this gene
results in congenital heart defects. To confirm this hypothesis,
further clinical and molecular biological studies are required.
Given that 1q21.1 duplication may not be well known
to clinical cardiologists, the present study will facilitate the
clinical recognition of 1q21.1 duplication in China. The results of
the high-resolution SNP array in combination with the detailed
phenotype analysis provided further evidence for the identification
of causative genes for CHD in the 1q21.1 duplication. The present
study may aid in the early diagnosis, genetic counseling and
effective long-term management of 1q21.1 duplication.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81200087, 81101475
and 81370204). The authors would like to thank the patient and her
family members for participating in the present study. The authors
would also like to thank the State Key Laboratory of Medical
Genetics of China for their technical assistance.
References
|
1
|
Digilio MC, Bernardini L, Consoli F, Lepri
FR, Giuffrida MG, Baban A, Surace C, Ferese R, Angioni A, Novelli
A, et al: Congenital heart defects in recurrent reciprocal 1q21.1
deletion and duplication syndromes: Rare association with pulmonary
valve stenosis. Eur J Med Genet. 56:144–149. 2013. View Article : Google Scholar
|
|
2
|
Dolcetti A, Silversides CK, Marshall CR,
Lionel AC, Stavropoulos DJ, Scherer SW and Bassett AS: 1q21.1
Microduplication expression in adults. Genet Med. 15:282–289. 2013.
View Article : Google Scholar
|
|
3
|
Mefford HC, Sharp AJ, Baker C, Itsara A,
Jiang Z, Buysse K, Huang S, Maloney VK, Crolla JA, Baralle D, et
al: Recurrent rearrangements of chromosome 1q21.1 and variable
pediatric phenotypes. New Engl J Med. 359:1685–1699. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Brunet A, Armengol L, Heine D, Rosell J,
García-Aragonés M, Gabau E, Estivill X and Guitart M: BAC array CGH
in patients with Velocardiofacial syndrome-like features reveals
genomic aberrations on chromosome region 1q21.1. BMC Med Genet.
10:1442009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Brunetti-Pierri N, Berg JS, Scaglia F,
Belmont J, Bacino CA, Sahoo T, Lalani SR, Graham B, Lee B, Shinawi
M, et al: Recurrent reciprocal 1q21.1 deletions and duplications
associated with microcephaly or macrocephaly and developmental and
behavioral abnormalities. Nat Genet. 40:1466–1471. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Christiansen J, Dyck JD, Elyas BG, Lilley
M, Bamforth JS, Hicks M, Sprysak KA, Tomaszewski R, Haase SM,
Vicen-Wyhony LM, et al: Chromosome 1q21.1 contiguous gene deletion
is associated with congenital heart disease. Circ Res.
94:1429–1435. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Guida V, Ferese R, Rocchetti M, Bonetti M,
Sarkozy A, Cecchetti S, Gelmetti V, Lepri F, Copetti M, Lamorte G,
et al: A variant in the carboxyl-terminus of connexin 40 alters GAP
junctions and increases risk for tetralogy of Fallot. Eur J Hum
Genet. 21:69–75. 2013. View Article : Google Scholar :
|
|
8
|
Soemedi R, Topf A, Wilson IJ, Darlay R,
Rahman T, Glen E, Hall D, Huang N, Bentham J, Bhattacharya S, et
al: Phenotype-specific effect of chromosome 1q21.1 rearrangements
and GJA5 duplications in 2436 congenital heart disease patients and
6760 controls. Hum Mol Genet. 21:1513–1520. 2012. View Article : Google Scholar :
|
|
9
|
Slavotinek AM: Novel microdeletion
syndromes detected by chromosome microarrays. Hum Genet. 124:1–17.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen JL, Yang YF, Huang C, Wang J, Yang JF
and Tan ZP: Clinical and molecular delineation of 16p13.3
duplication in a patient with congenital heart defect and multiple
congenital anomalies. Am J Med Genet A. 158A:685–688. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Huang C, Yang YF, Yin N, Chen JL, Wang J,
Zhang H and Tan ZP: Congenital heart defect and mental retardation
in a patient with a 13q33.1-34 deletion. Gene. 498:308–310. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tan ZP, Huang C, Xu ZB, Yang JF and Yang
YF: Novel ZFPM2/FOG2 variants in patients with double outlet right
ventricle. Clin Genet. 82:466–471. 2012. View Article : Google Scholar
|
|
13
|
Cooper GM, Coe BP, Girirajan S, Rosenfeld
JA, Vu TH, Baker C, Williams C, Stalker H, Hamid R, Hannig V, et
al: A copy number variation morbidity map of developmental delay.
Nat Genet. 43:838–846. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rosenfeld JA, Traylor RN, Schaefer GB,
McPherson EW, Ballif BC, Klopocki E, Mundlos S, Shaffer LG and
Aylsworth AS; 1q21.1 Study Group: Proximal microdeletions and
microduplications of 1q21.1 contribute to variable abnormal
phenotypes. Eur J Hum Genet. 20:754–761. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Greenway SC, Pereira AC, Lin JC, DePalma
SR, Israel SJ, Mesquita SM, Ergul E, Conta JH, Korn JM and
McCarroll SA: De novo copy number variants identify new genes and
loci in isolated sporadic tetralogy of Fallot. Nat Genet.
41:931–995. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Oliveira SM, Ehtisham J, Redwood CS,
Ostman-Smith I, Blair EM and Watkins H: Mutation analysis of
AMP-activated protein kinase subunits in inherited
cardiomyopathies: Implications for kinase function and disease
pathogenesis. J Mol Cell Cardiol. 35:1251–1255. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ganz T and Nemeth E: Hepcidin and
disorders of iron metabolism. Annu Rev Med. 62:347–360. 2011.
View Article : Google Scholar
|
|
18
|
Papanikolaou G, Samuels ME, Ludwig EH,
MacDonald ML, Franchini PL, Dubé MP, Andres L, MacFarlane J,
Sakellaropoulos N, Politou M, et al: Mutations in HFE2 cause iron
overload in chromosome 1q-linked juvenile hemochromatosis. Nat
Genet. 36:77–82. 2004. View
Article : Google Scholar
|
|
19
|
Olbrich H, Schmidts M, Werner C,
Onoufriadis A, Loges NT, Raidt J, Banki NF, Shoemark A, Burgoyne T,
Al Turki S, et al: Recessive HYDIN mutations cause primary ciliary
dyskinesia without randomization of left-right body asymmetry. Am J
Hum Genet. 91:672–684. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Menghsol SC, Harris RD and Ornvold K:
Thrombocytopenia and absent radii, TAR syndrome: Report of
cerebellar dysgenesis and newly identified cardiac and renal
anomalies. Am J Med Genet A. 123A:193–196. 2003. View Article : Google Scholar : PubMed/NCBI
|