Introduction
Ischemic heart disease is one of the most common
diseases worldwide. The traditional treatment of ischemic heart
disease includes the prevention of atherosclerosis, and
revascularization of the coronary arteries; however, these
strategies cannot reverse or repair myocardial necrosis. Heart
transplantation is an effective treatment for patients with
late-stage heart failure, however due to the insufficient supply of
organs there are limits to its clinical application (1–3).
Recently, the rapid development of stem cell technology has led to
novel treatment methods, including the transplantation of
mesenchymal stem cells (MSCs) to repair or regenerate damaged
myocardium (4–7). Previous studies have demonstrated
that MSCs transplanted into areas of myocardial ischemia may
differentiate into myocardial cells and repair necrotic myocardial
tissue. However, the effects of MSCs are insufficient, since the
majority of transplanted MSCs die shortly after transplantation in
the ischemic microenvironment (7,8).
Therefore, a key focus of research is to improve the survival of
MSCs following transplantation into ischemic tissue.
Prostaglandin E1 (PGE1), also
termed alprostadil, is an endogenous substance, which has numerous
effects, including vasodilation, protection of endothelial cells,
and inhibition of the activation and aggregation of neutrophils and
thrombocytes (9). Furthermore,
PGE1 is widely used in the treatment of ischemic heart
disease. Clinical research has previously demonstrated the
potential of PGE1 for improving myocardial
microcirculation, and counteracting the effects of
ischemia-reperfusion injury and apoptosis in the myocardium
(10–13). These findings indicate that PGE1
may have a general cytoprotective action; however, there are
currently no studies investigating whether PGE1 may
prevent apoptosis of MSCs.
Apoptosis is a type of physiological cell death, for
which it is considered difficult to generate a comprehensive in
vitro model. Serum deprivation (SD) injury in vitro is
widely used to mimic the ischemic environment (14,15).
The mitochondrial pathway is the major underlying mechanism of
physiological cell death in apoptosis (16,17),
and the Bcl-2 family proteins have an important role in the
apoptotic response (18,19). To the best of our knowledge, the
molecular mechanism by which PGE1 may inhibit apoptosis
of MSCs is currently unknown.
The present study established an in vitro
model of SD-induced apoptosis, in order to explore the potential
mechanisms by which PGE1 may improve the survival of
MSCs in the myocardial microenvironment following
transplantation.
Materials and methods
Animals
Sprague-Dawley rats (specific pathogen free; weight,
80–100 g) of either sex were purchased from the Animal Center of
Sun Yat-sen University (Guangzhou, China). All procedures of the
present study were approved by the Institutional Animal Care and
Use Committee of Sun Yat-sen University. The rats were maintained
in 12 h light/dark cycles at a temperature of 26–26°C and with a
humidity of 40–70%, with free access to a standard laboratory diet
and water.
Reagents and instruments
PGE1 was purchased from Zhuhai Schwarz
Pharma Co., Ltd. (Zhuhai, China). Dulbecco's modified Eagle's
medium (DMEM) and fetal bovine serum (FBS) were obtained from Gibco
Life Technologies (Carlsbad, CA, USA). Penicillin-streptomycin was
purchased from Solarbio Science & Technology Co., Ltd.
(Beijing, China). A Hoechst 33258 Staining kit and propidium iodide
(PI) were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Antibodies targeting Bcl-2 (cat. no. 2870), Bax (cat. no. 2772),
and caspase-3 (cat. no. 9662), were purchased from Cell Signaling
Technology Inc. (Danvers, MA, USA) and were used at a dilution of
1:1,000. A Bicinchoninic Acid (BCA) Protein Assay kit was purchased
from Kangchen Bio-tech (Shanghai, China). Enhanced
Chemiluminescence (ECL) solution was purchased from KeyGen Biotech
Co., Ltd. (Nanjing, China). Fluorescein isothiocyanate
(FITC)-labeled Annexin V and anti-rat CD90 (cat. no. 11-0900-81),
CD45 (cat. no. 17-0461-80), CD11b/c (cat. no. 12-0110-80), and CD29
(cat. no. 46-0291-80), antibodies were purchased from BD
Biosciences (San Jose, CA, USA).
Cell preparation and culture
MSCs were isolated from the femora and tibiae of
Sprague Dawley rats, which had were sacrificed by cervical
dislocation. The cells were cultured in DMEM supplemented with 10%
FBS and penicillin-streptomycin (50 U/ml), and were incubated at
37°C in a humidified atmosphere containing 5% CO2. The
culture medium was refreshed every 2–3 days. Each primary culture
was passaged 1:2 once the MSCs had grown to 80% confluence. MSCs at
passage 4, which were positive for CD90 and CD29, and negative for
CD45 and CD11b/c, were collected and used for subsequent
experiments.
SD-induced apoptosis
The MSCs were randomly divided into three groups and
cultured for 24 h. The control group was cultured with complete
medium supplemented with 10% FBS; the SD group was cultured with SD
medium; and the PGE1+SD group was cultured with SD
medium plus 10 ng/ml PGE1.
Hoechst 33342 staining
The culture medium was discarded, and the cells were
washed three times with phosphate-buffered saline (PBS), and fixed
in 4% paraformaldehyde for 15 min at room temperature. The fixing
solution was then discarded and the cells were washed with PBS for
5 min and incubated in Hoechst 33342 in the dark for 15 min at room
temperature. Following the 15 min incubation, the Hoechst 33342
solution was discarded, and the cells were washed for a further 5
min with PBS. The cells were then supplemented with PBS and
observed under an inverted fluorescence microscope (Olympus, Tokyo,
Japan). The apoptotic cells exhibited morphological changes,
including shrinkage and condensed nuclei.
Flow cytometric (FCM) analysis of
apoptosis
The MSCs from passage 4 were digested with 0.05%
Trypsin-EDTA (Gibco Life Technologies) for 5 min at 37°C,
resuspended at a concentration of 10×105 cells/ml and
centrifuged at 300 x g for 5 min. Following centrifugation, the
cells were collected in PBS at 4°C, washed twice, and resuspended
in 100 ml binding buffer (BD Biosciences). In a 5 ml dry flow tube
the cells were added to 5 µl Annexin V-FITC (BD Biosciences)
and 5 µl PI (BD Biosciences), lightly vortexed, and
incubated in the dark at room temperature for 15 min. Following
incubation, 400 µl binding buffer was added, and the flow
tube was placed on ice. PI and Annexin V-FITC fluorescence was
measured using a flow cytometer (BD FACSVerse; BD Biosciences;
excitation, 488 nm; emission, 615 nm). The research software (BD
FACSuite softwared; BD Bioscience) matched with FCM was used to
analyze the data. Positive Annexin V staining indicated apoptosis,
and positive PI staining indicated necrosis. The experiment was
repeated three times.
Western blot analysis of Bcl-2, Bax and
caspase-3
The MSCs were seeded into a 60 mm petri dish, at a
density of 10×105 cells/dish. The total protein was
extracted using radioimmunoprecipitation buffer (EMD Millipore),
supplemented with PMSF. The cells were sonicated briefly and
centrifuged at 10,000 x g at 4°C. The protein concentration was
measured using the BCA Protein Assay kit, according to the
manufacturer's instructions. Equal samples of protein (20
µg) were separated by 12% SDS-PAGE and then transferred onto
polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA,
USA). The membranes were incubated with blocking solution (Beyotime
Institute of Biotechnology, Jiangsu, China) at room temperature for
2 h, and then incubated with the following primary antibodies:
Bcl-2, Bax, caspase-3 and GAPDH (cat. no. KC-5G5; KangChen Biotech,
Shanghai, China; dilution, 1:10,000) at 4°C overnight. The
membranes were then incubated for 1 h at room temperature with the
appropriate horseradish peroxidase conjugated-secondary antibodies.
The blots were visualized using an enhanced chemiluminescence
solution (EMD Millipore) and were exposed to X-ray film (Kodak,
Tokyo, Japan). The density of the protein bands was analyzed using
ImageJ 1.41 software (National Institutes of Health, Bethesda, MD,
USA). The expression levels of the target proteins were normalized
to those of GAPDH.
Statistical analysis
The data are expressed as the mean ± standard
deviation. Comparisons between the groups were analyzed by one-way
analysis of variance or Student's t-test. Statistical analyses were
performed using SPSS 16.0 (SPSS, Inc., Chicago, IL, USA). P<0.05
was considered to indicate a statistically significant
difference.
Results
MSC morphology and identification
MSCs were isolated and expanded from Sprague Dawley
rats. The control MSCs were shown to sparsely attach to the culture
flasks, and the majority of cells displayed a spindle-like shape
(Fig. 1A). In the MSCs cultured
with SD medium cells did not display the typical spindle-like
shape, and a large number of floating dead cells were observed
(Fig. 1B). Following treatment
with PGE1, the morphology of the MSCs cultured with SD
changed, and the number of floating dead cells was reduced, as
compared with the cells cultured with SD alone (Fig. 1C). The MSCs at passage 4 were
stably positive for CD90 and CD29 markers, and negative for CD45
and CD11b/c markers.
PGE1 inhibits SD-induced
apoptosis
Hoechst 33342 staining showed that the majority of
the MSCs in the control group were round or oval-shaped, and had
light-blue regular nuclei (Fig.
2A). In the PGE1+SD group, the apoptotic cells were
round or oval-shaped, with bright-blue irregular nuclei, which
indicated chromosome condensation. The number of apoptotic cells in
the SD group was significantly increased, as compared with the
control group. Treatment with PGE1 significantly reduced
the rate of MSC apoptosis, as compared with the SD group.
Furthermore, flow cytometry of Annexin V/PI-stained cells was used
to quantify the apoptotic rate of the MSCs. The percentage of
apoptotic cells was significantly higher in the SD (P<0.01) and
SD+PGE1 (P<0.05) groups, as compared with the control
group (Fig. 2B and C). In
addition, the rate of apoptosis was lower in the
PGE1-treated MSCs, as compared with the untreated MSCs
cultured with SD (P<0.05).
 | Figure 2Apoptosis of MSCs exposed to serum
deprivation with or without PGE1 preconditioning. (A)
Morphological changes to the apoptotic cells were assessed by
Hoechst 33258 staining (magnification, ×100). Control, untreated
MSCs; SD, serum-deprived MSCs; SD+PGE1, serum-deprived
MSCs cultured with 10 ng/ml PGE1. MSCs were incubated
for 24 h. (B) Flow cytometric analysis of the influence of 10 ng/ml
PGE1 on SD-induced apoptosis. Apoptotic cells were
identified by Annexin V and PI staining; viable cells are Annexin
V−/PI−, early apoptotic cells are Annexin
V+/PI−, late apoptotic cells are Annexin
V+/PI+, and necrotic cells are Annexin
V−/PI+. (C) The results show that 10 ng/ml
PGE1 reduced apoptosis of MSCs after 24 h of serum
deprivation. The data are presented as the mean ± standard error
(n=5). **P<0.01, as compared with the control group;
#P<0.05, as compared with the SD group. MSC,
mesenchymal stem cells; Con, control; SD, serum deprived;
PGE1, prostaglandin E1; PI, propidium
iodide. |
SD influences the expression levels of
apoptosis-associated proteins Bax, Bcl-2 and caspase-3
The present study determined whether SD could affect
the expression levels of the proapoptotic Bcl-2 family members, Bax
and Bcl-2. Western blot analysis showed that the protein expression
levels of Bax were significantly increased and the protein
expression levels of Bcl-2 gradually decreased, following exposure
to SD for 12–24 h (Fig. 3A and B).
Cleaved caspase-3 expression was not detected in the control MSCs,
however its expression was significantly increased in the MSCs
cultured in SD medium (Fig.
3C).
Effects of PGE1 on the protein
expression levels of Bax, Bcl-2 and caspase-3
Treatment with PGE1 significantly reduced
SD-induced Bax protein expression levels and increased the protein
expression levels of Bcl-2 (Fig.
4A). PGE1 also reduced the protein expression levels
of cleaved caspase-3 in the MSCs (Figure 4B). These results indicate that
PGE1 was able to attenuate SD-induced apoptosis though
activation of Bax and deactivation of Bcl-2, thus reducing the
expression of cleaved caspase-3.
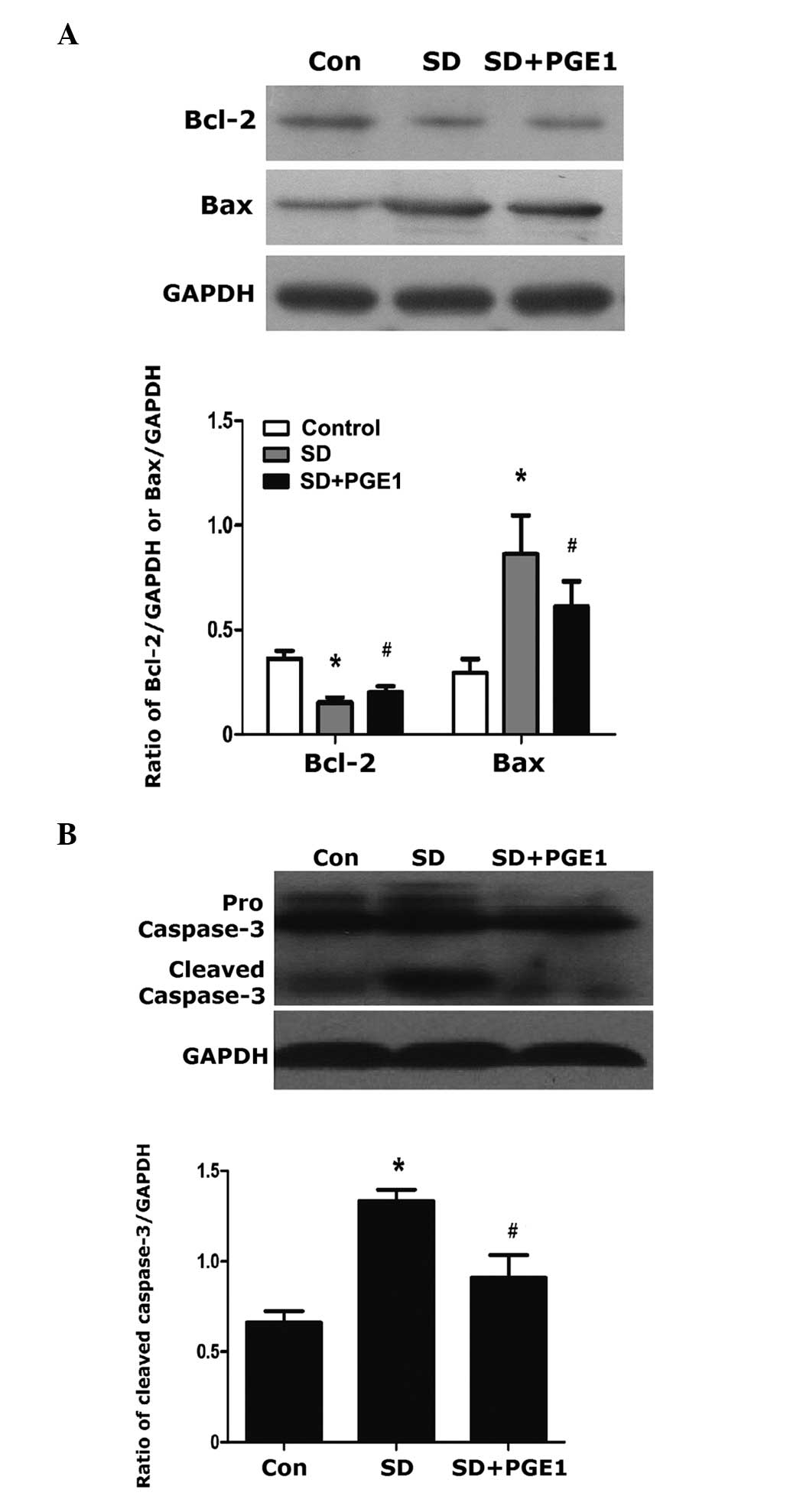 | Figure 4Western blot analysis of Bax, Bcl-2
and caspase-3 following 24 h of serum deprivation with or without
PGE1 preconditioning. Control, untreated MSCs; SD,
serum-deprived MSCs; SD+PGE1, serum-deprived MSCs
cultured with 10 ng/ml PGE1. MSCs were cultured at 37°C
in a humidified atmosphere containing 5% CO2. Protein
expression levels of (A) Bax and Bcl-2, and (B) caspase-3. GAPDH
was used as a loading control, and the expression levels of the
target proteins were determined relative to the levels of GAPDH.
Blots are shown from at least three independent experiments. The
data are presented as the mean ± standard error (n=5).
*P<0.01, as compared with the control group;
#P<0.05, ##P<0.01, as compared with the
SD group. MSC, mesenchymal stem cells; Con, control; SD, serum
deprived; PGE1, prostaglandin E. |
Discussion
The present study reported the protective effects of
PGE1 on SD-induced apoptosis in MSCs, and this effect
was shown to be mediated through the mitochondrial caspase-3
pathway. The results of the present study demonstrated that
treatment with PGE1 (10 ng/ml) decreased SD-induced
apoptosis in MSCs, as shown by Hoechst 33342 staining, flow
cytometry and measurement of caspase-3 protein expression levels.
Furthermore, PGE1 protected MSCs against SD-induced
apoptosis by downregulating Bax expression and upregulating Bcl-2
expression.
MSCs are nonhematogenic stem cells, which are
present in the bone marrow. Due to their availability, potential
for differentiation (20) and
amplification, and association with fewer ethical issues (21), MSCs have become a focus of
attention in the field of experimental and clinical research
regarding myocardial regeneration. However, the ischemic cardiac
microenvironment reduces the survival rate of transplanted cells,
and limits their therapeutic effects (22). More than 90% of MSCs have
previously been shown to die within 24 h of transplantation
(23). Another study demonstrated
that only ~21% of MSCs survive after 4 h of transplantation, and
only 3.6% survive seven days (24). To address the problem of poor
survival, research has focused on strategies that inhibit apoptosis
of MSCs and improve their therapeutic effects in the ischemic
myocardium. Such strategies include the use of genetically modified
stem cells, preconditioning of stem cells, and combination drug
therapy prior to transplantation of the cells into the damaged
myocardium (25). Genetic
modification (26,27) and preconditioning (28) of MSCs may improve the survival rate
of stem cells; however, these are difficult to perform clinically.
Whereas, combination drug therapy improves the viability of MSCs
and is convenient for clinical application. Zhang et al
(29) previously used rosuvastatin
as a combination therapy to improve the therapeutic efficacy of
MSCs for treating myocardial infarction. Furthermore, Dong et
al (30) used combination
therapy with atorvastatin, which was shown to activate
AMP-activated protein kinase (AMPK); phosphorylation of AMPK
resulted in activation of endothelial nitric oxide synthase. This
mechanism may also be associated with the protection of MSCs
against SD-induced apoptosis, through the mitochondrial apoptosis
signaling pathway.
PGE1 is widely used in the treatment of
ischemic heart disease. Previous research has identified the
ability of PGE1 to improve myocardial microcirculation,
reduce ischemicreperfusion injury, and exert anti-inflammatory and
antiapoptotic effects on the myocardium (10–13,31).
PGE1 also exhibits general cytoprotective effects and
anti-apoptotic activity (32). It
has previously been reported that PGE1 is able to
significantly upregulate antiapoptotic proteins, such as Bcl-2
(33); and downregulate Bax and
caspase-3 (13). The present study
established an in vitro SD-induced apoptosis model, in order
to explore the potential mechanisms for the protective effects of
PGE1 on MSCs. Apoptosis was detected by Hoechst 33258
and Annexin V-FITC/PI double staining. PGE1 was shown to
protect the MSCs against SD-induced apoptosis. However, apoptosis
involves a series of gene activation, expression and regulation;
therefore, further investigation is required to explore the
underlying molecular mechanisms by which PGE1 inhibits
apoptosis of MSCs.
The Bcl-2 family is an important
apoptosis-regulating family, which includes the antiapoptotic
molecule Bcl-2 and proapoptotic molecule Bax (34). Numerous studies have demonstrated
that Bcl-2 and Bax are associated with the mitochondrial membrane
(35–39). Bcl-2 is predominantly localized to
endoplasmic reticulum and mitochondrial membranes, where it
prevents the release of cytochrome c from the mitochondria
and inhibits glutathione leakage, thus blocking programmed cell
death (40,41). Bcl-2 can inhibit the activation of
caspases, including caspase-9 and caspase-3, and thereby acts as an
antiapoptotic agent (42). Bax is
predominantly localized to the cytosol, or may be loosely attached
to the mitochondrial membrane in an inactive form in healthy cells.
Apoptotic stimuli result in structural changes to Bax, which may
facilitate the translocation of Bax from the cytosol to the
mitochondria, leading to apoptosis (43). Bax exhibits extensive amino acid
homology with Bcl-2, and can form homodimers and heterodimers with
Bcl-2 in vivo (44). The
overexpression of Bax counteracts the death repressor activity of
Bcl-2, and the activation of caspase-3 is dependent on the ratio of
Bcl-2 to Bax, which controls cell survival and death following an
apoptotic stimulus (44,45). Caspase-3 is a critical mediator of
mitochondrial apoptosis (46),
which can be activated by SD in MSCs (47).
The effects of PGE1 on the inhibition of
caspase-3 and regulation of Bcl-2 and Bax have previously been
reported (13,33,48);
however, the present study is the first, to the best of our
knowledge, to report such effects in MSCs. To confirm these
findings, western blot analysis was used to detect the protein
expression levels of Bax, Bcl-2 and caspase-3. SD downregulated the
protein expression levels of Bcl-2, and upregulated the expression
levels of Bax in MSCs, resulting in overexpression of caspase-3,
which caused an increased rate of MSC apoptosis. Furthermore,
treatment with PGE1 significantly increased the
expression levels of Bcl-2 and inhibited the expression levels of
Bax and caspase-3, thereby attenuating apoptosis in MSCs. Apoptosis
is a complex process, and the SD model used in the present study
attempted to simulate the myocardial microenvironment in
vitro. However, further investigation is required to confirm
these findings in vivo.
In conclusion, the results of the present study
demonstrated that PGE1 exerts protective effects against
SD-induced MSC apoptosis. PGE1 downregulated the protein
expression levels of Bax and caspase-3, and upregulated the protein
expression levels of Bcl-2 in the SD in vitro model. These
findings may be useful in the clinical application of
PGE1 alongside MSC transplantation into ischemic tissue,
and may enhance the efficacy of cell therapy.
Acknowledgments
The authors of the present study would like to thank
Dr Run-min Guo, Professor Hong-fu Wu and Dr Qing-feng Xiang for
technical assistance. This study was supported by the program of
'One Hundred Talented Scholars' of Sun Yat-sen University (grant
no. F002009011).
References
|
1
|
Yu Q, Fan W and Cao F: Mechanistic
molecular imaging of cardiac cell therapy for ischemic heart
disease. Am J Physiol Heart Circ Physiol. 305:H947–H959. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Toyoda Y, Guy TS and Kashem A: Present
status and future perspectives of heart transplantation. Circ J.
77:1097–1110. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Burchill LJ and Ross HJ: Heart
transplantation in adults with end-stage congenital heart disease.
Future Cardiol. 8:329–342. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schächinger V, Erbs S, Elsässer A, et al:
Improved clinical outcome after intracoronary administration of
bone-marrow-derived progenitor cells in acute myocardial
infarction: final 1-year results of the REPAIR-AMI trial. Eur Heart
J. 27:2775–2783. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tse HF, Thambar S, Kwong YL, et al:
Prospective randomized trial of direct endomyocardial implantation
of bone marrow cells for treatment of severe coronary artery
diseases (PROTECT-CAD trial). Eur Heart J. 28:2998–3005. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Strauer BE and Steinhoff G: 10 years of
intracoronary and intra-myocardial bone marrow stem cell therapy of
the heart: from the methodological origin to clinical practice. J
Am Coll Cardiol. 58:1095–1104. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tang YL, Tang Y, Zhang YC, Qian K, Shen L
and Phillips MI: Improved graft mesenchymal stem cell survival in
ischemic heart with a hypoxia-regulated heme oxygenase-1 vector. J
Am Coll Cardiol. 46:1339–1350. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Menasché P: Current status and future
prospects for cell transplantation to prevent congestive heart
failure. Semin Thorac Cardiovasc Surg. 20:131–137. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhao XS, Pan W, Bekeredjian R and Shohet
RV: Endogenous endothelin-1 is required for cardiomyocyte survival
in vivo. Circulation. 114:830–837. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fang WT, Li HJ and Zhou LS: Protective
effects of prostaglandin E1 on human umbilical vein endothelial
cell injury induced by hydrogen peroxide. Acta Pharmacol Sin.
31:485–492. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Takikawa M, Sumi Y, Tanaka Y, et al:
Protective effect of pros-taglandin E1 on
radiation-induced proliferative inhibition and apoptosis in
keratinocytes and healing of radiation-induced skin injury in rats.
J Radiat Res. 53:385–394. 2012. View Article : Google Scholar
|
|
12
|
Li JH, Yang P, Li AL, Wang Y, Ke YN and Li
XL: Cardioprotective effect of liposomal prostaglandin E1 on a
porcine model of myocardial infarction reperfusion no-reflow. J
Zhejiang Univ Sci B. 12:638–643. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jia C, Dai C, Bu X, et al:
Co-administration of prostaglandin E1 with somatostatin attenuates
acute liver damage after massive hepatectomy in rats via inhibition
of inflammatory responses, apoptosis and endoplasmic reticulum
stress. Int J Mol Med. 31:416–422. 2013.
|
|
14
|
Zeng X, Yu SP, Taylor T, Ogle M and Wei L:
Protective effect of apelin on cultured rat bone marrow mesenchymal
stem cells against apoptosis. Stem Cell Res. 8:357–367. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chauvier D, Lecoeur H, Langonné A, et al:
Upstream control of apoptosis by caspase-2 in serum-deprived
primary neurons. Apoptosis. 10:1243–1259. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chalah A and Khosravi-Far R: The
mitochondrial death pathway. Adv Exp Med Biol. 615:25–45. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Suen DF, Norris KL and Youle RJ:
Mitochondrial dynamics and apoptosis. Genes Dev. 22:1577–1590.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Martinou JC and Youle RJ: Mitochondria in
apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev
Cell. 21:92–101. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gross A, McDonnell JM and Korsmeyer SJ:
BCL-2 family members and the mitochondria in apoptosis. Genes Dev.
13:1899–1911. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang X, Wang Y, Gao Y, et al: Maintenance
of high proliferation and multipotent potential of human hair
follicle-derived mesenchymal stem cells by growth factors. Int J
Mol Med. 31:913–921. 2013.PubMed/NCBI
|
|
21
|
Ding DC, Shyu WC and Lin SZ: Mesenchymal
stem cells. Cell Transplant. 20:5–14. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hale SL, Dai W, Dow JS and Kloner RA:
Mesenchymal stem cell administration at coronary artery reperfusion
in the rat by two delivery routes: a quantitative assessment. Life
Sci. 83:511–515. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hodgetts SI, Beilharz MW, Scalzo AA and
Grounds MD: Why do cultured transplanted myoblasts die in vivo? DNA
quantification shows enhanced survival of donor male myoblasts in
host mice depleted of CD4+ and CD8+ cells or Nk1.1+ cells. Cell
Transplant. 9:489–502. 2000.PubMed/NCBI
|
|
24
|
Tang YL, Tang Y, Zhang YC, Qian K, Shen L
and Phillips MI: Improved graft mesenchymal stem cell survival in
ischemic heart with a hypoxia-regulated heme oxygenase-1 vector. J
Am Coll Cardiol. 46:1339–1350. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mingliang R, Bo Z and Zhengguo W: Stem
cells for cardiac repair: status, mechanisms and new strategies.
Stem Cells Int. 2011:3109282011. View Article : Google Scholar
|
|
26
|
Huang J, Zhang Z, Guo J, et al: Genetic
modification of mesenchymal stem cells overexpressing CCR1
increases cell viability, migration, engraftment, and capillary
density in the injured myocardium. Circ Res. 106:1753–1762. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huang F, Zhu X, Hu XQ, et al: Mesenchymal
stem cells modified with miR-126 release angiogenic factors and
activate Notch ligand Delta-like-4, enhancing ischemic angiogenesis
and cell survival. Int J Mol Med. 31:484–492. 2013.
|
|
28
|
Liu XB, Chen H, Chen HQ, et al:
Angiopoietin-1 preconditioning enhances survival and functional
recovery of mesenchymal stem cell transplantation. J Zhejiang Univ
Sci B. 13:616–623. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang Z, Li S, Cui M, et al: Rosuvastatin
enhances the therapeutic efficacy of adipose-derived mesenchymal
stem cells for myocardial infarction via PI3K/Akt and MEK/ERK
pathways. Basic Res Cardiol. 108:3332013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dong Q, Yang Y, Song L, Qian H and Xu Z:
Atorvastatin prevents mesenchymal stem cells from hypoxia and
serum-free injury through activating AMP-activated protein kinase.
Int J Cardiol. 153:311–316. 2011. View Article : Google Scholar
|
|
31
|
Huang CL, Wu YW, Wang SS, et al:
Continuous intravenous infusion of prostaglandin E1 improves
myocardial perfusion reserve in patients with ischemic heart
disease assessed by positron emission tomography: a pilot study.
Ann Nucl Med. 25:462–468. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hara Y, Akamatsu Y, Maida K, et al: A new
liver graft preparation method for uncontrolled non-heart-beating
donors, combining short oxygenated warm perfusion and prostaglandin
E1. J Surg Res. 184:1134–1142. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ma XQ, Fu RF, Feng GQ, Wang ZJ, Ma SG and
Weng SA: Hypoxia-reoxygenation-induced apoptosis in cultured
neonatal rat cardiomyocyets and the protective effect of
prostaglandin E. Clin Exp Pharmacol Physiol. 32:1124–1130. 2005.
View Article : Google Scholar
|
|
34
|
Dietrich JB: Apoptosis and anti-apoptosis
genes in the Bcl-2 family. Arch Physiol Biochem. 105:125–135.
1997.In French. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Renault TT, Teijido O, Antonsson B, Dejean
LM and Manon S: Regulation of Bax mitochondrial localization by
Bcl-2 and Bcl-x(L): keep your friends close but your enemies
closer. Int J Biochem Cell Biol. 45:64–67. 2013. View Article : Google Scholar
|
|
36
|
Degli EM and Dive C: Mitochondrial
membrane permeabilisation by Bax/Bak. Biochem Biophys Res Commun.
304:455–461. 2003. View Article : Google Scholar
|
|
37
|
Nemec KN and Khaled AR: Therapeutic
modulation of apoptosis: targeting the BCL-2 family at the
interface of the mitochondrial membrane. Yonsei Med J. 49:689–697.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Autret A and Martin SJ: Emerging role for
members of the Bcl-2 family in mitochondrial morphogenesis. Mol
Cell. 36:355–363. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Garcia-Saez AJ, Fuertes G, Suckale J and
Salgado J: Permeabilization of the outer mitochondrial membrane by
Bcl-2 proteins. Adv Exp Med Biol. 677:91–105. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hockenbery D, Nuñez G, Milliman C,
Schreiber RD and Korsmeyer SJ: Bcl-2 is an inner mitochondrial
membrane protein that blocks programmed cell death. Nature.
348:334–336. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Haeberlein SL: Mitochondrial function in
apoptotic neuronal cell death. Neurochem Res. 29:521–530. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mukhopadhyay A, Shishodia S, Suttles J, et
al: Ectopic expression of protein-tyrosine kinase Bcr-Abl
suppresses tumor necrosis factor (TNF)-induced NF-kappa B
activation and IkappaBalpha phosphorylation. Relationship with
down-regulation of TNF receptors. J Biol Chem. 277:30622–30628.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Tan KO, Fu NY, Sukumaran SK, et al: MAP-1
is a mitochondrial effector of Bax. Proc Natl Acad Sci USA.
102:14623–14628. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Oltvai ZN, Milliman CL and Korsmeyer SJ:
Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that
accelerates programmed cell death. Cell. 74:609–619. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Heon Seo K, Ko HM, Kim HA, et al:
Platelet-activating factor induces up-regulation of antiapoptotic
factors in a melanoma cell line through nuclear factor-kappaB
activation. Cancer Res. 66:4681–4686. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lakhani SA, Masud A, Kuida K, et al:
Caspases 3 and 7: key mediators of mitochondrial events of
apoptosis. Science. 311:847–851. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhu W, Chen J, Cong X, Hu S and Chen X:
Hypoxia and serum deprivation-induced apoptosis in mesenchymal stem
cells. Stem Cells. 24:416–425. 2006. View Article : Google Scholar
|
|
48
|
Liu HJ, Ma JW, Qiao ZY and Xu B: Study of
molecular mechanism of Prostaglandin E1 in inhibiting coronary
heart disease. Mol Biol Rep. 40:6701–6708. 2013. View Article : Google Scholar : PubMed/NCBI
|
















