Introduction
Alzheimer's disease (AD) is one of the most common
and complex neurodegenerative disorders and is characterized by a
progressive decline of memory and cognition (1). The disease is defined by specific
neuropathological changes of neurofibrillary tangles (NFT) and
amyloid plaques that accumulate in vulnerable brain regions
(2,3). Neurodegeneration in the development
of AD varies substantially across cell types and regions. Of note,
it has been demonstrated that hippocampal CA1 pyramidal neurons are
particularly vulnerable to neurodegeneration and bear NFTs during
the early stages of AD (4,5); however, the underlying mechanisms of
their degeneration have remained elusive.
AD is thought to be caused by the dysregulation of a
large number of genes and the consequent alteration of their
complex interactions, which finally contributes to the broad
spectrum of disease phenotypes (6–9).
Microarray technology, which provides researchers with a tool to
assess the expression levels of thousands of genes simultaneously,
offers the possibility of gaining insight into gene networks
disturbed in intricate human disease such as AD, and to obtain
possible molecular clues regarding the underlying mechanisms of the
pathophysiology of AD. Previous studies have used this technique to
more comprehensively enhance the knowledge of the cellular and
molecular changes underlying AD (10–14).
Although these studies have yielded significant novel insights,
inconsistencies are present across these studies due to limitations
based on small sample sizes and various results obtained by
different groups with different laboratory protocols, microarray
platforms and microarray data interpretations (15). In view of this, the present study
integrated hippocampus gene expression datasets from multiple AD
microarray studies to overcome these limitations of individual
studies, resolve inconsistencies and provide significant novel
insight into the complex biological processes involved in AD.
Materials and methods
Identification of eligible gene
expression profiles of hippocampi of patients with AD
Hippocampal gene expression profiling studies in
patients with AD were identified by searching the Gene Expression
Omnibus database (GEO; http://www.ncbi.nlm.nih.gov/geo) (16). The following key words and their
combinations were used: 'Alzheimer's disease', 'hippocampus', 'gene
expression' and 'microarray.' Only experimental studies that had
performed hippocampal gene expression profiling in patients with AD
as well as normal control (NC) subjects were used. Non-human
studies, review articles and integrated analyses of expression
profiles were excluded.
Data preprocessing
Normalization is crucial for comparing different
microarray datasets. The heterogeneity caused by different
microarray platforms, gene nomenclature and clinical samples may
make it difficult to compare the expression data directly. However,
inappropriate normalization may contribute to the skewing of
results and reduce their statistical significance. Consequently, a
global normalization approach to minimize any inconsistencies
should be included. For this propose, MATLAB Bioinformatics Toolbox
was used in the present study to pre-process the raw microarray
data of each study by Quantile normalization and log2
transformation to obtain intensity values.
Statistical analysis
MATLAB software, version 2013a (MathWorks, Natick,
MA) was used to identify the differently expressed probe sets in
the hippocampal tissues of patients with AD compared to those of NC
subjects. A gene-specific t-test was performed, followed by
calculation of the P-value and the effect size of the individual
microarray study. Fisher's combined probability method was used to
combine P-values from multiple studies, and the random effects
model was used to combine effect sizes from multiple studies. Genes
with an effect size >0.8 and a P-value <0.01 were selected as
the significantly differentially expressed genes (DEGs).
Functional annotation of DEGs
To gain insight into the biological functions of
DEGs, gene ontology (GO) classification was performed. GO provides
a common descriptive framework as well as functional annotation and
classification for analyzing the gene expression datasets.
Furthermore, Kyoto Encyclopedia of Genes and Genomes (KEGG;
http://www.genome.jp/kegg/) pathway
enrichment analysis was performed to map the potential pathways of
the DEGs. The KEGG pathway database is a recognized and
comprehensive database, which includes an extensive variety of
biochemical pathways (17). The
online-based software GENECODIS, version 3 was utilized in the
present analysis (18).
Protein-protein interactions (PPIs)
network construction
PPI analysis allows for the assessment of protein
functions at the molecular level, which are divided into the
categories of cellular growth, development, metabolism,
differentiation and apoptosis (19). The detection of key
protein-interacting ions in the PPI networks of AD is important for
the interpretation of cellular regulatory mechanisms in the
development of the disease (20).
The present study adopted the Search Tool for the Retrieval of
Interacting Genes/Proteins (http://www.string-db.org/), a database of known and
predicted protein interactions, to construct the PPI network and
then visualized the distribution characteristics of the top 10 up-
and downregulated DEGs in the network with Cytoscape software,
version 3.2.0 (21).
Results
Identification of DEGs in hippocampi of
patients with AD
The present study collected a total of four datasets
of gene expression profiles in hippocampi of patients with AD
according to the inclusion criteria; in total, data on the gene
expression in 73 samples from patients with AD and 61 samples from
control subjects were analyzed. The studies containing the
individual hippocampal expression profiles in patients with AD are
listed in Table I (12,22–24).
A total of 11,494 genes from four expression profiling studies were
assessed. For the purpose of global normalization, the raw
microarray data were pre-processed by Quantile normalization and
log2 transformation to obtain intensity values for each probe,
which were used in the gene expression profiling. Subsequently,
MATLAB software was utilized to identify DEGs in hippocampi between
patients with AD and control subjects. Finally, a total of 295 DEGs
were regarded as significantly differentially expressed between
samples of patients with AD and NC subjects (109 upregulated and
186 downregulated genes) when the threshold was set as P<0.01
and effect size >0.8. A list of the top 10 most significantly
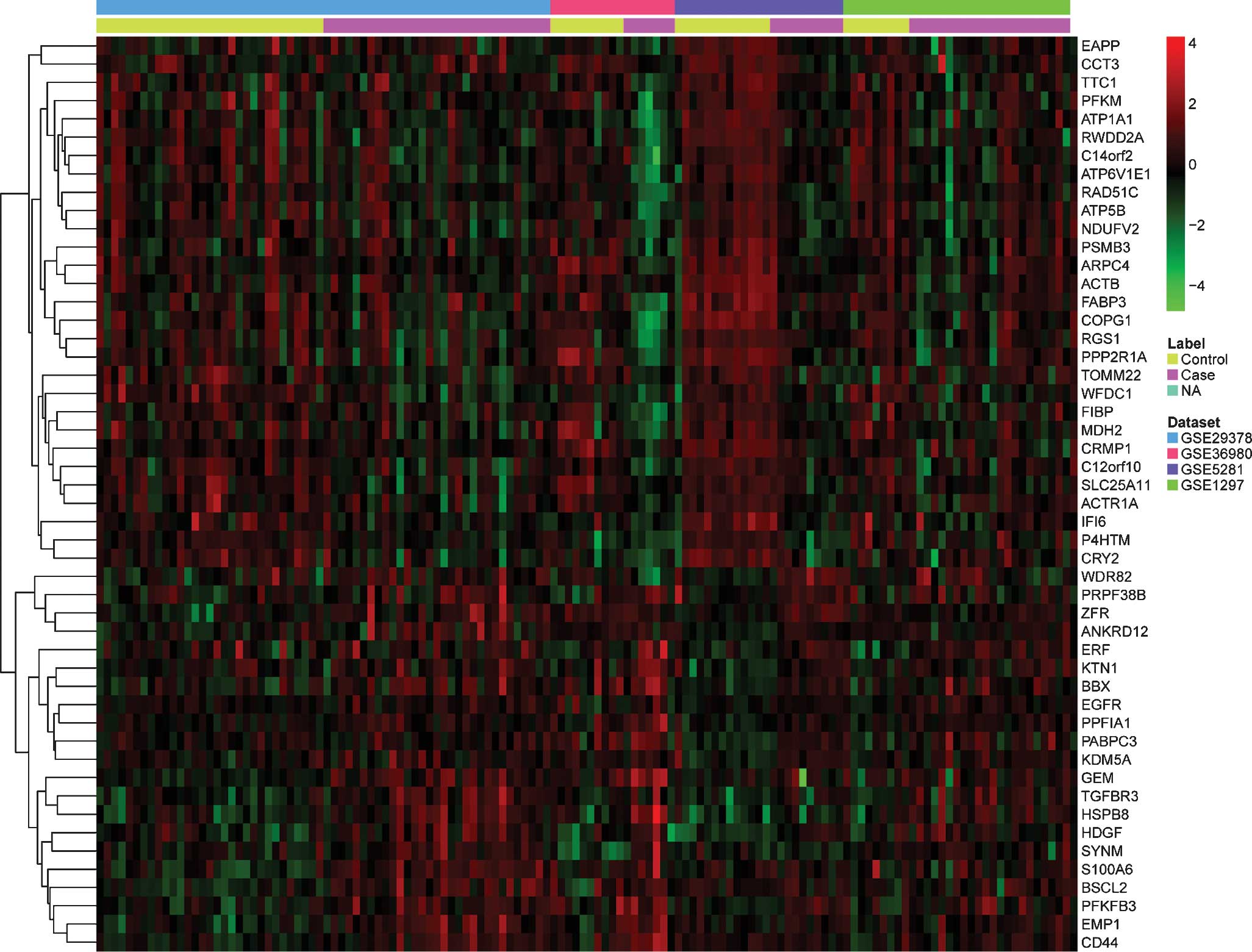
up- or downregulated genes is presented in Table II. The pattern of expressional
changes of the top 50 most significantly DEGs is displayed in a
heat map in Fig. 1.
 | Table ICharacteristics of the individual
studies. |
Table I
Characteristics of the individual
studies.
| GEO ID | Platform | Samples (n)
(cases:controls) | Country | Year | Author |
|---|
| GSE29378 | GPL6947 Illumina
HumanHT-12
V3.0 expression beadchip | 31:32 | USA | 2013 | Miller JA (22) |
| GSE36980 | GPL6244
[HuGene-1_0-st]
Affymetrix Human Gene 1.0 ST Array | 7:10 | Japan | 2013 | Hokama M (23) |
| GSE5281 | GPL570
[HG-U133_Plus_2] Affymetrix
Human Genome U133 Plus 2.0 Array | 13:10 | USA | 2007 | Liang WS (24) |
| GSE1297 | GPL96 [HG-U133A]
Affymetrix
Human Genome U133A Array | 22:9 | USA | 2004 | Blalock EM (12) |
| Table IITop 10 most significantly up- or
down-regulated differentially expressed genes. |
Table II
Top 10 most significantly up- or
down-regulated differentially expressed genes.
| Gene ID | Gene symbol | Official full
name | P-value | Effect size |
|---|
| Upregulated
genes |
| 51663 | ZFR | Zinc finger RNA
binding protein |
1.12×10−6 | 0.94853 |
| 2669 | GEM | GTP binding protein
overexpressed in skeletal muscle |
1.21×10−6 | 1.1503 |
| 6277 | S100A6 | S100 calcium binding
protein A6 |
2.74×10−6 | 1.0918 |
| 80335 | WDR82 | WD repeat domain
82 |
3.05×10−6 | 0.80325 |
| 5209 | PFKFB3 |
6-Phosphofructo-2-kinase/fructose-2,
6-Biphosphatase 3 |
3.24×10−6 | 1.1908 |
| 3895 | KTN1 | Kinectin 1 (kinesin
receptor) |
3.36×10−6 | 1.1643 |
| 3068 | HDGF | Hepatoma-derived
growth factor (high-mobility group protein 1-like) |
4.57×10−6 | 1.1580 |
| 2077 | ERF | Ets2 repressor
factor |
5.16×10−6 | 1.0677 |
| 7049 | TGFBR3 | Transforming growth
factor, beta receptor III |
5.34×10−6 | 1.1818 |
| 5042 | PABPC3 | Poly(A) binding
protein, cytoplasmic 3 |
7.09×10−6 | 1.0283 |
| Downregulated
genes |
| 22820 | COPG1 | Coatomer protein
complex, subunit gamma |
8.69×10−8 | 1.3017 |
| 58189 | WFDC1 | WAP four-disulfide
core domain 1 |
4.19×10−7 | 1.3844 |
| 10093 | ARPC4 | Tubulin tyrosine
ligase-like family, member 3; actin related protein 2/3 complex,
subunit 4, 20kDa |
7.43×10−7 | 1.1458 |
| 60 | ACTB | Actin, beta |
1.08×10−6 | 1.1657 |
| 9158 | FIBP | fibroblast growth
factor (acidic) intracellular binding protein |
1.15×10−6 | 1.1794 |
| 56993 | TOMM22 | Translocase of
outer mitochondrial membrane 22 homolog (yeast) |
2.35×10−6 | 1.0773 |
| 2537 | IFI6 | Interferon,
alpha-inducible protein 6 |
2.57×10−6 | 1.1754 |
| 9556 | C14orf2 | Chromosome 14 open
reading frame 2 |
2.72×10−6 | 1.1734 |
| 55837 | EAPP | E2F-associated
phosphoprotein |
3.11×10−6 | 1.0270 |
| 5889 | RAD51C | RAD51 homolog C
(S. cervisiae) |
5.16×10−6 | 1.0461 |
The upregulated gene with the lowest P-value was
ZFR, which is mainly expressed in neural tissue, but also weakly
expressed in other tissue types (25,26),
suggesting a neuronal function. A recent study identified ZFR as a
putative genes associated with hereditary spastic paraplegias by
using whole-exome sequencing (27). The downregulated gene with the
lowest P-value was COPG1, whose function has yet to be
determined.
Functional annotation
To investigate the biological roles of the DEGs in
the hippocampi of patients with AD, the present study performed a
categorized GO enrichment analysis. GO provides a common
descriptive framework and functional annotation of the gene
datasets. GO categories are separated into three groups: Biological
processes, cellular components and molecular function. The present
study examined GO categories separately using the web-based
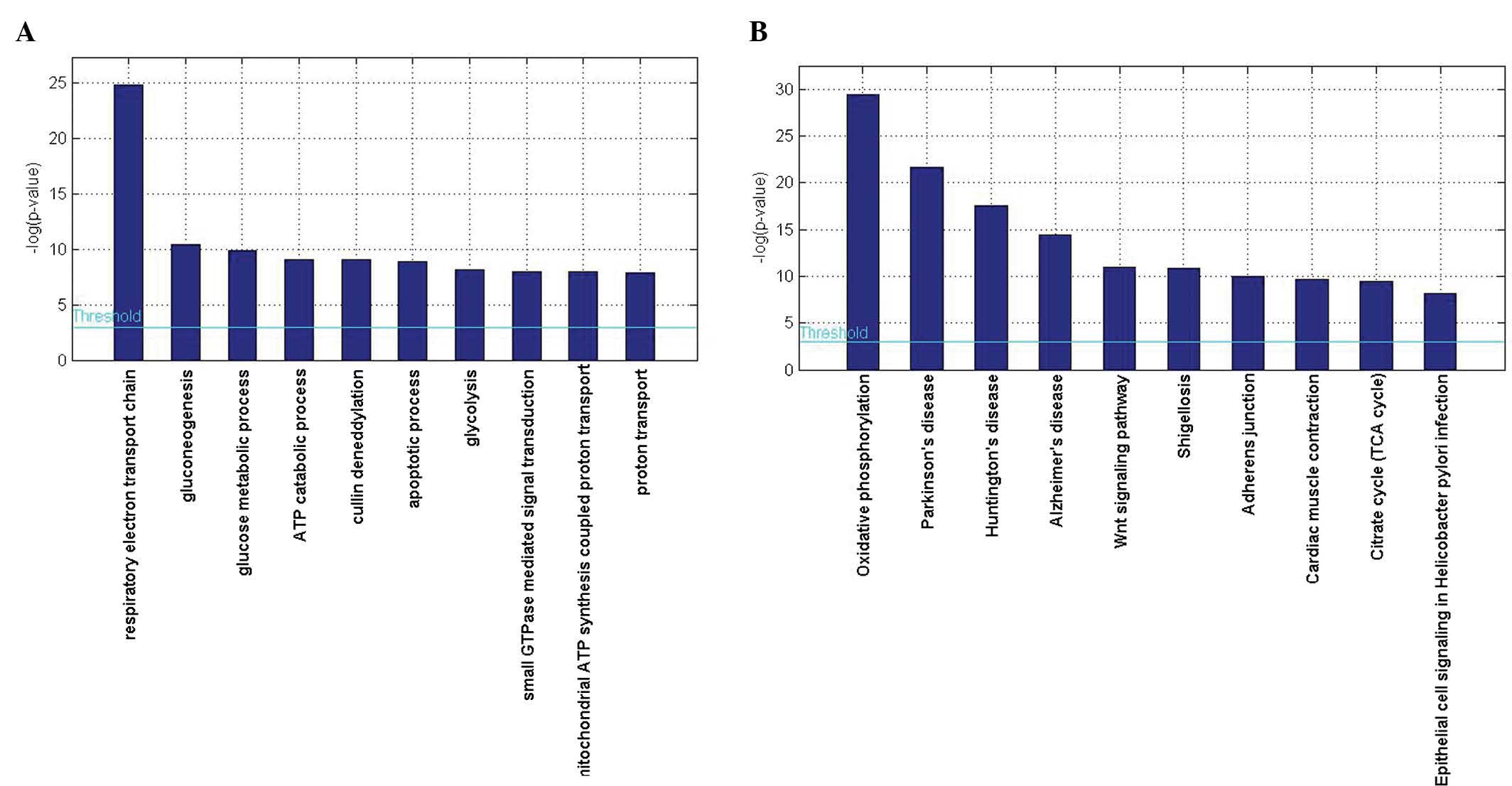
software GENECODIS. The results showed that genes associated with
the respiratory electron transport chain (GO: 0022904;
P=1.64×10−11) and gluconeogenesis (GO: 0006094;
P=2.84×10−5) were significantly enriched among
biological processes, while for molecular functions, protein
binding (GO:0005515; P=3.03×10−29) and nucleotide
binding (GO: 0000166; P=5.41×10−14) were significantly
enriched, and with regard to cellular components, genes associated
with the cytoplasm (GO:0005737; P=8.67×10−33) and
mitochondrion (GO: 0005739; P=1.00×10−23) were
significantly enriched (Table
III, Fig. 2A).
| Table IIIGO terms of differentially expressed
genes (top 15). |
Table III
GO terms of differentially expressed
genes (top 15).
| GO ID | GO term | No. of genes | FDR |
|---|
| Biological
processes |
| GO:0022904 | Respiratory
electron transport chain | 14 |
1.64×10−11 |
| GO:0006094 |
Gluconeogenesis | 7 |
2.84×10−5 |
| GO:0006006 | Glucose metabolic
process | 9 |
4.94×10−5 |
| GO:0006200 | ATP catabolic
process | 8 |
1.15×10−4 |
| GO:0010388 | Cullin
deneddylation | 4 |
1.18×10−4 |
| GO:0006915 | Apoptotic
process | 19 |
1.35×10−4 |
| GO:0006096 | Glycolysis | 6 |
2.91×10−4 |
| GO:0007264 | Small GTPase
mediated signal transduction | 13 |
3.44×10−4 |
| GO:0042776 | Mitochondrial ATP
synthesis coupled proton transport | 4 |
3.61×10−4 |
| GO:0015992 | Proton
transport | 6 |
3.84×10−4 |
| GO:0007165 | Signal
transduction | 26 |
7.19×10−4 |
| GO:0006120 | Mitochondrial
electron transport, NADH to ubiquinone | 5 |
1.01×10−3 |
| GO:0006810 | Transport | 17 |
1.26×10−3 |
| GO:0048146 | Positive regulation
of fibroblast proliferation | 5 |
1.30×10−3 |
| GO:0016071 | mRNA metabolic
process | 10 |
1.40×10−3 |
| Molecular
function |
| GO:0005515 | Protein
binding | 117 |
3.03×10−29 |
| GO:0000166 | Nucleotide
binding | 59 |
5.41×10−14 |
| GO:0005524 | ATP binding | 37 |
5.48×10−7 |
| GO:0005525 | GTP binding | 16 |
1.32×10−5 |
| GO:0046961 | Proton-transporting
ATPase activity, rotational mechanism | 5 |
4.49×10−5 |
| GO:0003924 | GTPase
activity | 12 |
4.55×10−5 |
| GO:0015631 | Tubulin
binding | 5 |
8.56×10−5 |
| GO:0046933 | Hydrogen ion
transporting ATP synthase activity, rotational mechanism | 4 |
3.08×10−4 |
| GO:0005509 | Calcium ion
binding | 18 |
5.87×10−4 |
| GO:0008137 | NADH dehydrogenase
(ubiquinone) activity | 5 |
6.24×10−4 |
| GO:0003713 | Transcription
coactivator activity | 10 |
6.81×10−4 |
| GO:0022857 | Transmembrane
transporter activity | 5 |
8.83×10−4 |
| GO:0005516 | Calmodulin
binding | 8 |
1.17×10−3 |
| GO:0047485 | Protein N-terminus
binding | 6 |
1.81×10−3 |
| GO:0003878 | ATP citrate
synthase activity | 2 |
1.87×10−3 |
| Cellular
components |
| GO:0005737 | Cytoplasm | 133 |
8.67×10−23 |
| GO:0005739 | Mitochondrion | 60 |
1.00×10−23 |
| GO:0005829 | Cytosol | 62 |
6.03×10−16 |
| GO:0005743 | Mitochondrial inner
membrane | 25 |
1.38×10−15 |
| GO:0005634 | Nucleus | 101 |
4.71×10−14 |
| GO:0005856 | Cytoskeleton | 26 |
7.29×10−7 |
| GO:0005625 | Soluble
fraction | 17 |
1.64×10−6 |
| GO:0016020 | Membrane | 66 |
3.04×10−6 |
| GO:0005886 | Plasma
membrane | 60 |
3.94×10−6 |
| GO:0005759 | Mitochondrial
matrix | 12 |
4.18×10−6 |
| GO:0005654 | Nucleoplasm | 25 |
4.28×10−6 |
| GO:0045121 | Membrane raft | 9 |
3.28×10−5 |
| GO:0005730 | Nucleolus | 31 |
4.98×10−5 |
| GO:0005753 | Mitochondrial
proton-transporting ATP synthase complex | 4 |
1.58×10−4 |
| GO:0030054 | Cell junction | 15 |
2.50×10−4 |
The present study subsequently performed a KEGG
pathway enrichment analysis in order to further evaluate the
biological roles of the DEGs. A hypergeometric test with P<0.05
was used as the criterion for pathway detection. According to the
KEGG analysis, oxidative phosphorylation was the most significant
pathway (P=1.61×10−13). Furthermore, pathways involved
in Parkinson's disease (P=3.77×10−10) and Huntington's
disease (P=2.26×10−8) were also highly enriched
(Table IV, Fig. 2B).
| Table IVKEGG pathways of differentially
expressed genes (top 15). |
Table IV
KEGG pathways of differentially
expressed genes (top 15).
| KEGG ID | KEGG term | No. of genes | FDR | Genes |
|---|
| hsa00190 | Oxidative
phosphorylation | 17 |
1.61×10−13 | COX4I1, NDUFAB1,
UQCRC1, NDUFV2, COX6A1, COX6B1, ATP5L, ATP5J2, ATP6AP1, ATP6V1E1,
NDUFA4, ATP5C1, NDUFA9, ATP6V1B2, NDUFS3, UQCRH, ATP5B |
| hsa05012 | Parkinson's
disease | 14 |
3.77×10−10 | COX4I1, NDUFAB1,
VDAC2, UQCRC1, NDUFV2, COX6A1, COX6B1, NDUFA4, ATP5C1, NDUFA9,
UCHL1, NDUFS3, UQCRH, ATP5B |
| hsa05016 | Huntington's
disease | 14 |
2.26×10−8 | COX4I1, NDUFAB1,
VDAC2, UQCRC1, NDUFV2, COX6A1, COX6B1, EP300, NDUFA4, ATP5C1,
NDUFA9, NDUFS3, UQCRH, ATP5B |
| hsa05010 | Alzheimer's
disease | 12 |
5.59×10−7 | COX4I1, NDUFAB1,
UQCRC1, NDUFV2, COX6A1, COX6B1, NDUFA4, ATP5C1, NDUFA9, NDUFS3,
UQCRH, ATP5B |
| hsa04310 | Wnt signaling
pathway | 10 |
1.70×10−5 | TCF7L1, PORCN,
EP300, PPP2R1A, JUN, CTNNBIP1, NFAT5, TBL1X, RBX1, DAAM1 |
| hsa05131 | Shigellosis | 7 |
1.97×10−5 | ARPC4, NFKBIA,
ELMO1, ACTB, ARPC1A, WASF2, CD44 |
| hsa04520 | Adherens
junction | 7 |
4.80×10−5 | TCF7L1, ACTB,
EP300, EGFR, WASF2, FYN, SORBS1 |
| hsa04260 | Cardiac muscle
contraction | 7 |
6.66×10−5 | COX4I1, UQCRC1,
COX6A1, COX6B1, ATP1A1, SLC9A6, UQCRH |
| hsa00020 | Citrate cycle (TCA
cycle) | 5 |
7.87×10−5 | ACLY, OGDHL, MDH2,
ACO2, SUCLG1 |
| hsa05120 | Epithelial cell
signaling in Helicobacter pylori infection | 6 |
2.87×10−4 | NFKBIA, ATP6AP1,
ATP6V1E1, EGFR, JUN, ATP6V1B2 |
| hsa03050 | Proteasome | 5 |
3.05×10−4 | PSMD13, PSMB3,
PSMC1, PSMC3, PSMD8 |
| hsa05130 | Pathogenic
Escherichia coli infection | 5 |
1.11×10−3 | ARPC4, ACTB,
ARPC1A, TUBA1B, FYN |
| hsa00250 | Alanine, aspartate
and glutamate metabolism | 4 |
1.64×10−3 | NIT2, GAD1, ADSL,
GOT1 |
| hsa05100 | Bacterial invasion
of epithelial cells | 5 |
3.27×10−3 | ARPC4, ELMO1, ACTB,
ARPC1A, WASF2 |
| hsa04810 | Regulation of actin
cytoskeleton | 8 |
4.03×10−3 | ARPC4, SSH3,
MYL12B, ACTB, ARPC1A, EGFR, WASF2, ITGB8 |
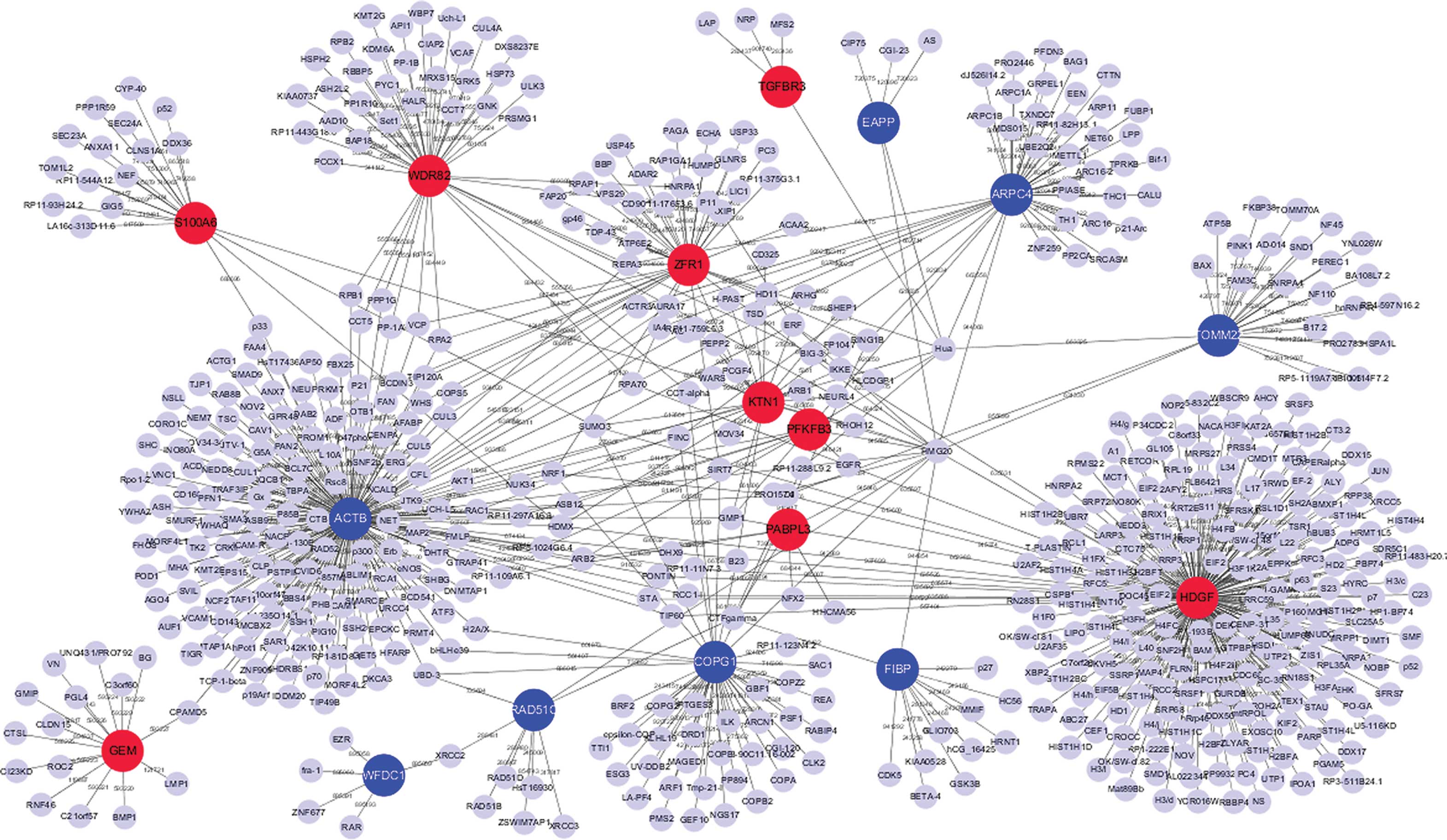
PPI network construction
The present study established the PPI networks of
the top 10 upregulated and downregulated DEGs using Cytoscape
software. The interaction network included 863 nodes and 1,304
edges. In the PPI network, degrees of interaction were defined to
determine the number of neighbors a node directly connected to, and
nodes with a high degree of interaction were defined as hub
proteins. The significant hub proteins included β-actin (ACTB;
degree, 268), hepatoma-derived growth factor (HDGF; degree, 218)
and WD repeat-containing protein 82 (WDR82; degree, 87) (Fig. 3).
Discussion
The present study aimed to identify altered
hippocampus gene expression and their further association with
other biological processes that regulate causative factors for AD
to provide diagnostic factors or therapeutic targets of AD. An
integrated analysis of DEGs from four publicly available GEO
datasets of hippocampi from patients with AD was performed. In
total, 295 genes were consistently differentially expressed across
the studies with 109 upregulated genes and 186 downregulated genes.
The upregulated gene with the lowest P-value was ZFR, which is
mainly expressed in neural tissue (25,26)
and may therefore have a role in neuronal function. A recent study
identified ZFR as a putative gene associated with hereditary
spastic paraplegias by using whole-exome sequencing (27), and from this, the present study
deduced that ZFR may be implicated in the underlying processes of
AD, which is required to be confirmed by further experiments. The
downregulated gene with the lowest P-value was COPG1, whose
function remains to be elucidated.
In line with previous studies, certain genes
identified in the present study have been closely associated with
the development of AD, including S100A6 and TGFBR3. A study on the
roles of S100 family proteins in nervous system function and
disease found that mRNA expression levels of six family members
(S100A1, S100B, S100A6, S100A10, S100A4, S100A13) displayed a
100-fold range in mouse brains, five of which (S1100A1, S100A6,
S100A10, S100A13, and S100B) showed age-dependent increases in
adult mice that ranged from 5- to 20-fold (28). S100A6-protein immunoreactivity was
found to be specifically located within astrocytes associated to
amyloid plaques in an APP/London transgenic mouse model of AD, as
well as in the brains of patients with AD. S100A6 was upregulated
in the amygdala as well as in hippocampal regions (29). Another study detected that biglycan
proteoglycans were upregulated in familial AD, while TGFBR3 was
markedly downregulated in sporadic AD fibroblasts. Furthermore, the
differential expression of TGFBR3 in familial AD and sporadic AD
cells was associated with the severity of AD (30).
In the present study,the results of the PPI network
analysis of the top 10 upregulated and downregulated DEGs indicated
that the significant hub proteins included ACTB, HDGF and WDR82.
ACTB, which encodes β-actin, is a candidate reference gene for
normalization of target gene expression in polymerase chain
reaction (PCR) analysis due to its high conservation. A previous
study determined the mRNA levels of ACTB and other genes in the
frontal cortex of patients with AD and control subjects using PCR
analysis with SYBR Green technology to identify suitable endogenous
reference genes in human post-mortem brain tissues for the
expression analysis of potential candidate genes associated with AD
(31); according to this study,
ACTB was the least suitable candidate with reliable expression
among a set of suitable endogenous reference genes due to low
expression stability in the frontal cortex of AD (32). Of note, the actin cytoskeleton has
been reported to have an important role in AD pathology by
mediating synaptic degeneration (32).
In order to elucidate the biological roles of the
DEGs in AD, a categorized GO enrichment analysis was performed in
the present study. The results showed that the respiratory electron
transport chain was the most significantly enriched GO category for
biological processes. To further evaluate the biological role for
the DEGs, the present study performed a KEGG pathway enrichment
analysis. According to the KEGG analysis, the most significantly
enriched pathway was oxidative phosphorylation. A previous study
provided evidence of neuronal metabolic impairments at the
transcriptomic and protein level in the brains of patients with AD
(33), which was ascribed to the
downregulation of mitochondria-associated genes, in particular,
oxidative phosphorylation genes in consistency with the fact that
AD is a degenerative disease. Furthermore, the present study found
that the pathways of several neurodegenerative diseases, including
Parkinson's disease, Huntington's disease and Alzheimer's disease,
were also highly enriched according to the KEGG pathway enrichment
analysis, which was due to dysregulation of genes associated with
mitochondrial energy metabolism, including COX4I1, NDUFAB1, UQCRC1,
NDUFV2, COX6A1, COX6B1. This finding validated the integrated
analysis methods used in the present study.
It is noteworthy that the present study had several
limitations. The heterogeneity of the datasets used may have
distorted the analysis, as clinical samples may have been
heterogeneous with regard to clinical activity or gender.
Furthermore, the effects of varying degrees of severity of AD on
the differences in hippocampal gene expression were not taken into
account. However, the present integrated analysis of different
datasets of hippocampal gene expression in patients with AD may
have facilitated the detection of genes that would have been missed
in the analysis of a single patient or study cohort. Despite these
limitations, the present study provided novel information regarding
the molecular mechanisms of AD; however, further analyses are
required to confirm the present findings.
In conclusion, the present study performed an
integrated analysis, which provided significant insight into the
global molecular changes associated with AD pathology. Furthermore,
the present study identified DEGs as well as other biological
functions, which may contribute to the successful identification of
diagnostic factors or therapeutic targets for AD and the
development of effective targeted therapies. Further functional
studies may provide additional insight into the role of the DEGs in
the pathophysiology of AD.
References
|
1
|
Hommet C, Mondon K, Constans T, Beaufils
E, Desmidt T, Camus V and Cottier JP: Review of cerebral
microangiopathy and Alzheimer's disease: Relation between white
matter hyperintensities and microbleeds. Dement Geriatr Cogn
Disord. 32:367–378. 2011. View Article : Google Scholar
|
|
2
|
Hyman BT, Van Hoesen GW, Damasio AR and
Barnes CL: Alzheimer's disease: Cell-specific pathology isolates
the hippocampal formation. Science. 225:1168–1170. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kordower JH, Chu Y, Stebbins GT, DeKosky
ST, Cochran EJ, Bennett D and Mufson EJ: Loss and atrophy of layer
II entorhinal cortex neurons in elderly people with mild cognitive
impairment. Ann Neurol. 49:202–213. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Scheff SW, Price DA, Schmitt FA, DeKosky
ST and Mufson EJ: Synaptic alterations in CA1 in mild Alzheimer
disease and mild cognitive impairment. Neurology. 68:1501–1508.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kerchner GA, Hess CP, Hammond-Rosenbluth
KE, Xu D, Rabinovici GD, Kelley DA, Vigneron DB, Nelson SJ and
Miller BL: Hippocampal CA1 apical neuropil atrophy in mild
Alzheimer disease visualized with 7-T MRI. Neurology. 75:1381–1387.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
O'Brien RJ and Wong PC: Amyloid precursor
protein processing and Alzheimer's disease. Annu Rev Neurosci.
34:185–204. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Noble W, Hanger DP, Miller CC and
Lovestone S: The importance of tau phosphorylation for
neurodegenerative diseases. Front Neurol. 4:832013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Martin L, Latypova X, Wilson CM,
Magnaudeix A, Perrin ML, Yardin C and Terro F: Tau protein kinases:
Involvement in Alzheimer's disease. Ageing Res Rev. 12:289–309.
2013. View Article : Google Scholar
|
|
9
|
Liu CC, Kanekiyo T, Xu H and Bu G:
Apolipoprotein E and Alzheimer disease: Risk, mechanisms and
therapy. Nat Rev Neurol. 9:106–118. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Auld DS, Kornecook TJ, Bastianetto S and
Quirion R: Alzheimer's disease and the basal forebrain cholinergic
system: Relations to beta-amyloid peptides, cognition and treatment
strategies. Prog Neurobiol. 68:209–245. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Beckmann L, Fischer C, Deck KG, Nolte IM,
te Meerman G and Chang-Claude J: Exploring haplotype sharing
methods in general and isolated populations to detect gene(s) of a
complex genetic trait. Genet Epidemiol. 21(Suppl 1): S554–S559.
2001.
|
|
12
|
Blalock EM, Geddes JW, Chen KC, Porter NM,
Markesbery WR and Landfield PW: Incipient Alzheimer's disease:
Microarray correlation analyses reveal major transcriptional and
tumor suppressor responses. Proc Natl Acad Sci USA. 101:2173–2178.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Colangelo V, Schurr J, Ball MJ, Pelaez RP,
Bazan NG and Lukiw WJ: Gene expression profiling of 12633 genes in
Alzheimer hippocampal CA1: Transcription and neurotrophic factor
down-regulation and up-regulation of apoptotic and pro-inflammatory
signaling. J Neurosci Res. 70:462–473. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mufson EJ, Counts SE and Ginsberg SD: Gene
expression profiles of cholinergic nucleus basalis neurons in
Alzheimer's disease. Neurochem Res. 27:1035–1048. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Siddiqui AS, Delaney AD, Schnerch A,
Griffith OL, Jones SJ and Marra MA: Sequence biases in large scale
gene expression profiling data. Nucleic Acids Res. 34:e832006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Barrett T, Wilhite SE, Ledoux P,
Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH,
Sherman PM, Holko M, et al: NCBI GEO: Archive for functional
genomics data sets-update. Nucleic Acids Res. 41:D991–D995. 2013.
View Article : Google Scholar
|
|
17
|
Altermann E and Klaenhammer TR:
PathwayVoyager: Pathway mapping using the Kyoto Encyclopedia of
Genes and Genomes (KEGG) database. BMC Genomics. 6:602005.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tabas-Madrid D, Nogales-Cadenas R and
Pascual-Montano A: GeneCodis3: A non-redundant and modular
enrichment analysis tool for functional genomics. Nucleic Acids
Res. 40:W478–W483. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Giot L, Bader JS, Brouwer C, Chaudhuri A,
Kuang B, Li Y, Hao YL, Ooi CE, Godwin B, Vitols E, et al: A protein
interaction map of Drosophila melanogaster. Science. 302:1727–1736.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Schaefer MH, Lopes TJ, Mah N, Shoemaker
JE, Matsuoka Y, Fontaine JF, Louis-Jeune C, Eisfeld AJ, Neumann G,
Perez-Iratxeta C, et al: Adding protein context to the human
protein-protein interaction network to reveal meaningful
interactions. PLoS Comput Biol. 9:e10028602013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Miller JA, Woltjer RL, Goodenbour JM,
Horvath S and Geschwind DH: Genes and pathways underlying regional
and cell type changes in Alzheimer's disease. Genome Med. 5:482013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hokama M, Oka S, Leon J, Ninomiya T, Honda
H, Sasaki K, Iwaki T, Ohara T, Sasaki T, LaFerla FM, et al: Altered
expression of diabetes-related genes in Alzheimer's disease brains:
The Hisayama study. Cereb Cortex. 24:2476–2488. 2014. View Article : Google Scholar :
|
|
24
|
Liang WS, Dunckley T, Beach TG, Grover A,
Mastroeni D, Walker DG, Caselli RJ, Kukull WA, McKeel D, Morris JC,
et al: Gene expression profiles in anatomically and functionally
distinct regions of the normal aged human brain. Physiol Genomics.
28:311–322. 2007. View Article : Google Scholar
|
|
25
|
Meagher MJ, Schumacher JM, Lee K,
Holdcraft RW, Edelhoff S, Disteche C and Braun RE: Identification
of ZFR, an ancient and highly conserved murine
chromosome-associated zinc finger protein. Gene. 228:197–211. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kleines M, Gärtner A, Ritter K and Schaade
L: Cloning and expression of the human single copy homologue of the
mouse zinc finger protein zfr. Gene. 275:157–162. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Novarino G, Fenstermaker AG, Zaki MS,
Hofree M, Silhavy JL, Heiberg AD, Abdellateef M, Rosti B, Scott E,
Mansour L, et al: Exome sequencing links corticospinal motor neuron
disease to common neurodegenerative disorders. Science.
343:506–511. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zimmer DB, Chaplin J, Baldwin A and Rast
M: S100-mediated signal transduction in the nervous system and
neurological diseases. Cell Mol Biol (Noisy-le-grand). 51:201–214.
2005.
|
|
29
|
Boom A, Pochet R, Authelet M, Pradier L,
Borghgraef P, Van Leuven F, Heizmann CW and Brion JP: Astrocytic
calcium/zinc binding protein S100A6 over expression in Alzheimer's
disease and in PS1/APP transgenic mice models. Biochim Biophys
Acta. 1742:161–168. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bellucci C, Lilli C, Baroni T, Parnetti L,
Sorbi S, Emiliani C, Lumare E, Calabresi P, Balloni S and Bodo M:
Differences in extracellular matrix production and basic fibroblast
growth factor response in skin fibroblasts from sporadic and
familial Alzheimer's disease. Mol Med. 13:542–550. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Leduc V, Legault V, Dea D and Poirier J:
Normalization of gene expression using SYBR green qPCR: A case for
paraoxonase 1 and 2 in Alzheimer's disease brains. J Neurosci
Methods. 200:14–19. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bamburg JR and Bloom GS: Cytoskeletal
pathologies of Alzheimer disease. Cell Motil Cytoskeleton.
66:635–649. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liang WS, Reiman EM, Valla J, Dunckley T,
Beach TG, Grover A, Niedzielko TL, Schneider LE, Mastroeni D,
Caselli R, et al: Alzheimer's disease is associated with reduced
expression of energy metabolism genes in posterior cingulate
neurons. Proc Natl Acad Sci USA. 105:4441–4446. 2008. View Article : Google Scholar : PubMed/NCBI
|