Introduction
Non-alcoholic fatty liver disease (NAFLD) is a cause
of fatty liver, occurring when fat is deposited in the liver
(steatosis) not due to excessive alcohol use. It is associated with
insulin resistance and metabolic syndrome (1). NAFLD is currently considered to be
the most common cause of chronic liver disease worldwide (2) and associated with other potentially
life-threatening diseases and increased mortality from
cardiovascular diseases, malignancy and hepatic complications.
NAFLD has also been found to be associated with several
extra-hepatic disorders, including breast cancer, polycystic ovary
syndrome and renal dysfunction (3–15).
NAFLD encompasses a wide spectrum of liver diseases ranging from
simple steatosis to non-alcoholic steatohepatitis (NASH) (1), which is the most extreme form of
NAFLD and is regarded as a major cause of cirrhosis of the liver of
unknown cause (16). NASH is a
major health problem and complicated by portal hypertension and
hepatic decompensation, and is occasionally accompanied with
hepatocellular carcinoma (HCC) (17).
Recently, various treatment modalities have been
applied in NASH, including lifestyle modification, surgical
intervention and pharmacological agents (including insulin
sensitizers, anti-oxidant agents, lipid-lowering agents and tumor
necrosis factor-alpha (TNF-α) antagonists) (18–20).
However, to date, there are no US Food and Drug
Administration-approved medical therapies for NASH or liver
fibrosis. There is an urgent requirement for novel therapeutic
approaches (17,21). As inflammatory activation has a
significant role in NASH progression, anti-inflammatory therapy for
NASH is of increasing interest (22). For example, TNF-α antagonist
pentoxifylline, interleukin (IL)-6 antagonist Sant7 and the
TNF-α-specific monoclonal antibodies infliximab, adalimumab and
certolizumab have been studied in a number of clinical NAFH trials
(23). At present,
anti-inflammatory strategies for NASH are restricted to targeting
one single cytokine, e.g., IL-1 receptor, IL-6 or TNF-α. However,
multiple cytokines are involved in the inflammatory response of
NASH. Therefore, targeting an upstream signaling molecule that
regulates multiple cytokine production may improve the objective
response rates (24). Recently, a
novel neural pathway termed as cholinergic anti-inflammatory
reflex, has been discovered, which inhibits the production of
inflammatory cytokines and may be a novel anti-inflammatory
strategy for NASH.
The α7-nicotinic acetylcholine receptor (α7 nAChR)
is a sub-type of nicotinic acetylcholine receptor and has a crucial
role in mediating the cholinergic anti-inflammatory signaling
pathway (25). It is expressed on
different types of cells, including neurons, macrophages,
lymphocytes, monocytes and dendritic cells. Activation of the α7
nAChR expressed on resident macrophages may suppress the local
inflammation by reducing the production of pro-inflammatory
cytokines TNF-α and IL-6, which are closely associated with certain
inflammatory diseases, including sepsis, rheumatoid arthritis,
asthma and diabetes (26). It is
therefore indicated that α7 nAChR is a promising target for
developing novel anti-inflammatory drugs. However, to date, it has
remained to be clarified whether α7 nAChR is associated with
NASH.
The present study assessed whether activation of α7
nAChR was able to prevent the progression of NASH, and whether
targeting of α7 nAChR may represent a novel strategy for NASH
therapy.
Materials and methods
Cell culture and reagents
RAW 264.7 cells were obtained from the American Type
Culture Collection (Manassas, VA, USA) and maintained in Dulbecco's
modified Eagle's medium (Gibco-BRL, Invitrogen Life Technologies,
Carlsbad, CA, USA) supplemented with 10% heat-inactivated fetal
bovine serum (Invitrogen Life Technologies) in a humidified
atmosphere of 95% air with 5% CO2 at 37°C. Nicotine was
purchased from Sigma-Aldrich (St. Louis, MO, USA; n=80).
Experimental protocols and animals
C57 male mice at four weeks of age (weight, 17–23 g)
were purchased from the Model Animal Research Center of Nanjing
University (Nanjing, China) and housed in the laboratory animal
center of Zhejiang Chinese Medical University (Hangzhou, China) at
22°C with a 12-h light/dark cycle. Mice were randomly divided into
four groups (n=10) and fed either a control diet (10% kcal as fat;
Mediscience Ltd., Yangzhou, China) or a high-fat diet (HFD; 60%
kcal as fat; Medicience Ltd) for 18 weeks with or without nicotine
for three weeks: 1) Control group, mice were fed a control diet and
supplemented with normal saline; (2) HFD group, mice were fed a HFD and
supplemented with normal saline; (3) control + nicotine 5 mg/kg group, mice
were fed a control diet and supplemented with nicotine at a dose of
5 mg/kg; (4) HFD + nicotine 5
mg/kg group, mice were fed a HFD and supplemented with nicotine at
the dose of 5 mg/kg. During the 18 weeks of feeding, the body
weight was measured every week. At the end of the experiment, mice
were sacrificed by cardiac puncture under CO2
anesthesia, and livers were collected for further analysis.
All animals used in the present study were housed
and cared for in accordance with the Chinese Pharmacological
Society Guidelines for Animal Use. The protocols of the present
study were approved by the Committee on the Ethics of Animal
Experiments of the Zhejiang Chinese Medical University (Hangzhou,
China; permit no. 2012-1849). All surgeries were performed under
sodium pentobarbital anesthesia (70 mg/kg; Sigma-Aldrich) and all
efforts were made to minimize suffering.
Biochemical serum analysis
The activity levels of aspartate aminotransferase
(AST) and alanine aminotransferase (ALT) (10) were determined using an automatic
blood chemical analyzer (Dry-Chem 4000i; Fujifilm, Tokyo, Japan).
TNF-α and IL-6 levels were measured using ELISA kits (cat. nos.
EK0527 and EK0441; Boster Biological Inc., Wuhan, China).
Histological examination and Oil Red O
staining
The fixed liver tissue was cut into 3-mm blocks,
which were embedded in paraffin and cut into 4-µm slices.
After being de-paraffinized using xylene and ethanol dilutions and
re-hydration, the sections were stained with hematoxylin and eosin
(H&E; Bogoo, Shanghai, China) to examine the tissue structure,
inflammatory cell infiltration, necrosis and lipid
accumulation.
For Oil Red O staining, cryosections of optimal
cutting temperature compound-embedded liver tissues (10 mm) were
fixed in 10% buffered formalin for 5 min at room temperature,
stained with Oil Red O (Biohao Company, Wuhan, China) for 1 h,
washed with 10% isopropanol and then counterstained with
hematoxylin for 30 sec. A Nikon E600 microscope (Nikon, Tokyo,
Japan) and Leica Application Suite (Leica Microsystems, Inc.,
Buffalo Grove, IL, USA) were used to capture images of the Oil Red
O-stained tissue sections at 40× magnification.
Isolation of macrophages from liver
tissue
Forty normal, healthy mice were anesthetized and
liver tissues were perfused in situ via the superior vena
cava with a perfusion buffer (13 Hanks' balanced salt solution;
Gino Biological Medical Technology, Co., Ltd., Hangzhou, China),
followed by a digestion buffer [13 Hanks' balanced salt solution,
supplemented with 0.05% collagenase (Type IV; Sigma-Aldrich), 1.25
mmol/l CaCl2, 4 mmol/l MgSO4 and 10 mmol/l
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid]. The resulting
cell suspension was filtered through a sterile 100-mm nylon mesh
(Solarbio, Beijing, China) and centrifuged at 50 ×g to selectively
sediment hepatocytes from non-parenchymal cells (NPCs). The pellet
of hepatocytes was re-suspended and subsequently washed two more
times with centrifugation at 50 ×g. The NPCs in the first and
second supernatants from the low-speed centrifugations were
pelleted by high-speed centrifugation (1,300 × g), followed by
re-suspension in a small volume prior to isopycnic sedimentation in
Percoll as previously described (27). Cell viability (90%) was determined
by trypan blue exclusion (Sigma-Aldrich). The Kupffer cells were
treated with lipopolysaccharide (LPS; Escherichia coli
O111:B4; Sigma-Aldrich; 100 nM) for 16–18 h, following which the
culture medium was replaced with medium without serum, and in the
presence or absence of nicotine (concentrations between 0 and 10
µm) for 6 h. Treatment with α-bungarotoxin (α-BGT; Zhongxin
Dongtai Company, Laiyang, China) was also performed.
Western blot analysis
Whole-cell lysates were prepared using
radioimmunoprecipitation assay lysis buffer (Beyotime Institute of
Biotechnology, Nantong, China), protein concentrations were
detected using a bicinchoninic acid assay kit (Beyotime Institute
of Biotechnology) and western blotting was performed, as previously
described (27). Briefly, equal
amounts of protein were separated by SDS-PAGE. Proteins were then
transferred onto nitrocellulose membranes and identified with
anti-α7 nAChR polyclonal antibody (cat. no. 23791-AP; Proteinch
USA), anti-NF-κB monoclonal antibody (cat. no. 4764S; Cell
Signaling Technology, Inc., Beverly, MA, USA), anti-inhibitor of
NF-κB (IκB) antibody (cat. no. 4814; CST Company, Boston, MA, USA),
anti-extracellular signal-regulated kinase (ERK) monoclonal
antibody (cat. no. 20G11; Cell Signaling Technology, Inc.) and
anti-GAPDH antibodies (Santa Cruz Biotechnology, Inc., Dallas, TX,
USA) at 1:1,000. Detection was performed using a horseradish
peroxidase-conjugated secondary antibody and SuperSignal West Pico
Chemiluminescent Substrate (Pierce Biotechnology, Inc., Rockford,
IL, USA) according to the manufacturer's instructions. Kodak films
(Kodak, Rochester, NY, USA) were used to visualize the gels.
Statistical analysis
Values are expressed as the mean ± standard
deviation. Statistical analyses were performed using one-way
analysis of variance or the unpaired Student's t-test as
indicated. Statistical analysis was performed using SPSS v.10.0
statistical software (SPSS, Inc., Chicago, IL, USA). P<0.05 was
considered to indicate a statistically significant difference
between values.
Results
HFD-induced NASH
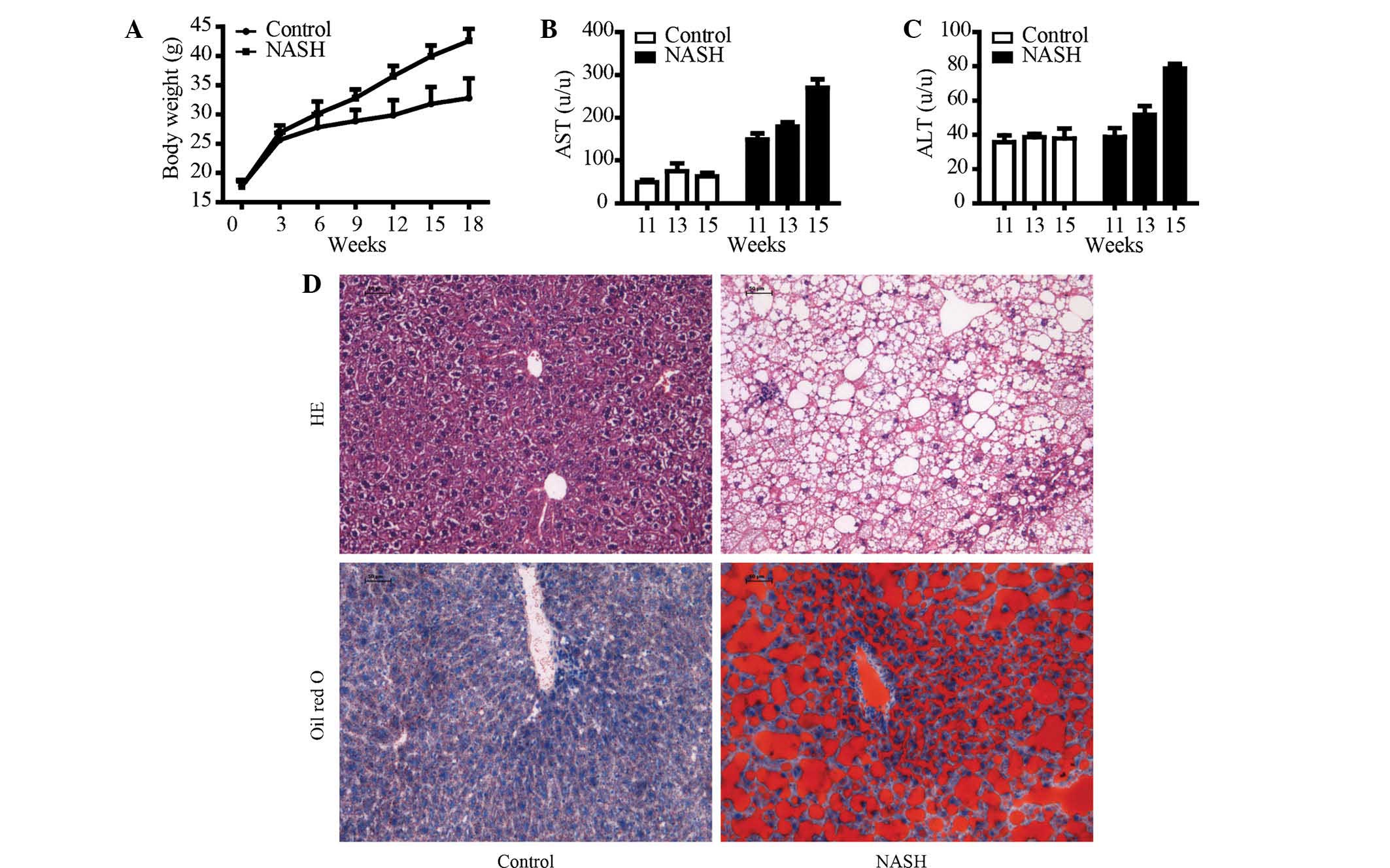
In the present study, a mouse model of NASH was
generated by intake of a HFD. After 18 weeks of HFD intake, the
body weight was significantly increased, which indicated the
establishment of the obesity mouse model (Fig. 1A). As shown in Fig. 2B and C, activities of AST and ALT
were increased in mice on an HFD compared with those in the control
mice which received a normal diet. It appeared that the HFD induced
liver injury. To determine whether HFD induced hepatic steatosis,
liver pathological examination by H&E staining was performed
(Fig. 1D). The hepatic cell
structure in the control group was normal. However, the HFD
increased hepatic damage with obvious hepatic necrosis. Further
examination of the hepatic lipid accumulation status with Oil Red O
staining revealed that the HFD significantly induced hepatic lipid
accumulation compared to that in the control group.
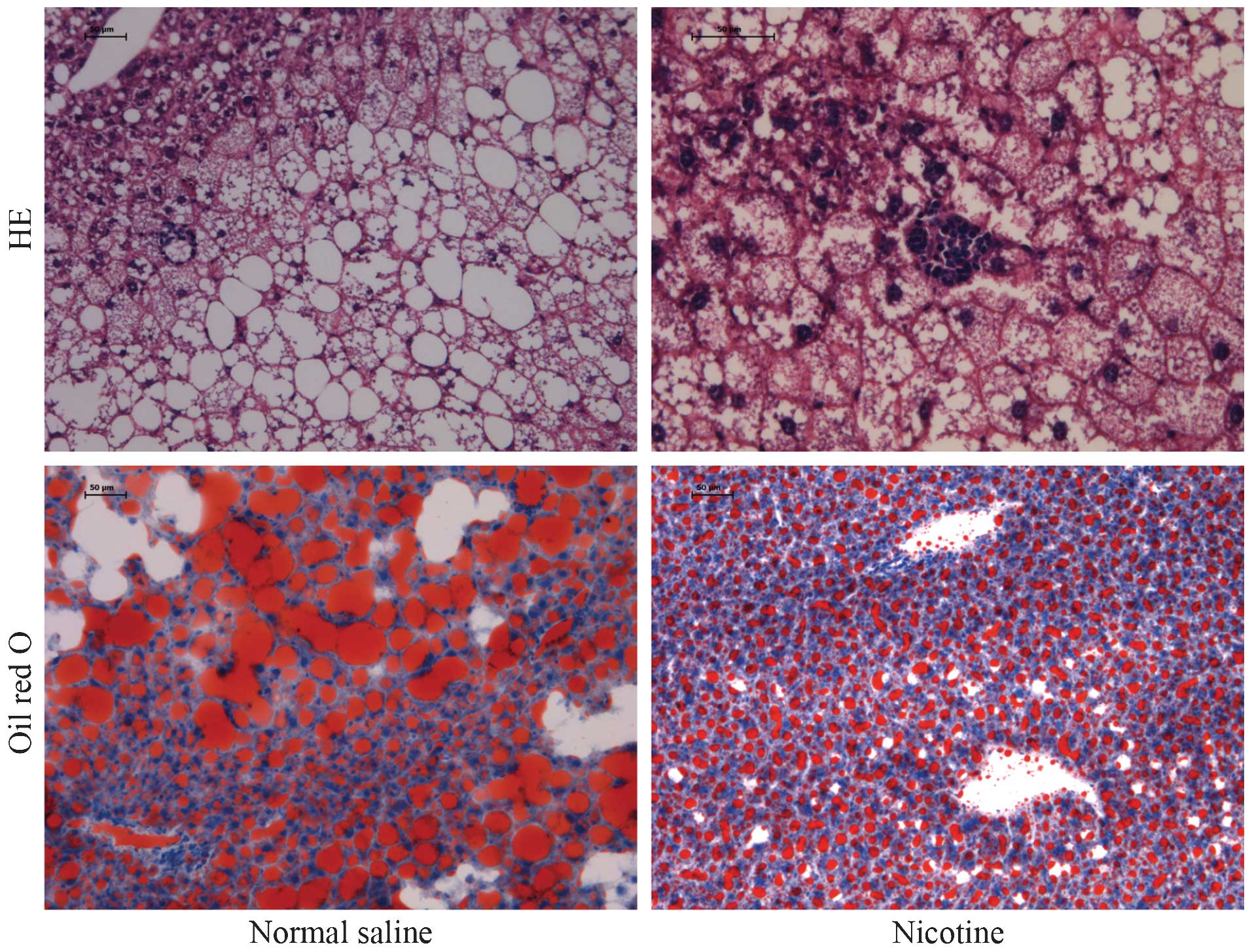
Activation of α7 nAChR attenuates
HFD-induced hepatic steatosis
In order to identify whether activation of α7 nAChR
can prevent NASH and the subsequent hepatic injury, α7 nAChR
agonist nicotine was administered to mice receiving the HFD. As
shown in Fig. 2, administration of
nicotine significantly, but not completely, prevented HFD-induced
hepatic necrosis and hepatic lipid accumulation.
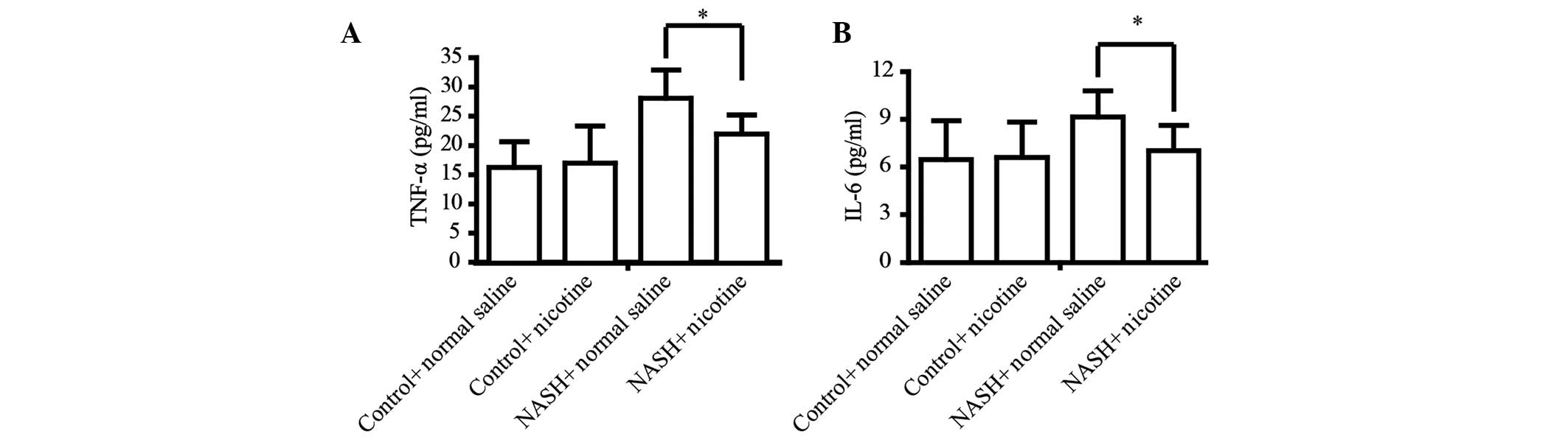
Activation of α7 nAChR attenuates
HFD-induced hepatic inflammation
Inflammation is the main pathological consequence of
HFD-induced NASH and is characterized by a release of inflammatory
factors, which contributes to hepatic fibrosis (28,29).
Thus, the present study determined whether nicotine can prevent
HFD-induced hepatic inflammation. The secretion of the classic
inflammatory factors TNF-α and IL-6 was detected by ELISA. The HFD
significantly upregulated the serum levels of TNF-α and IL-6 in
mice. However, nicotine treatment significantly attenuated
HFD-induced upregulation of serum TNF-α and IL-6 (Fig. 3).
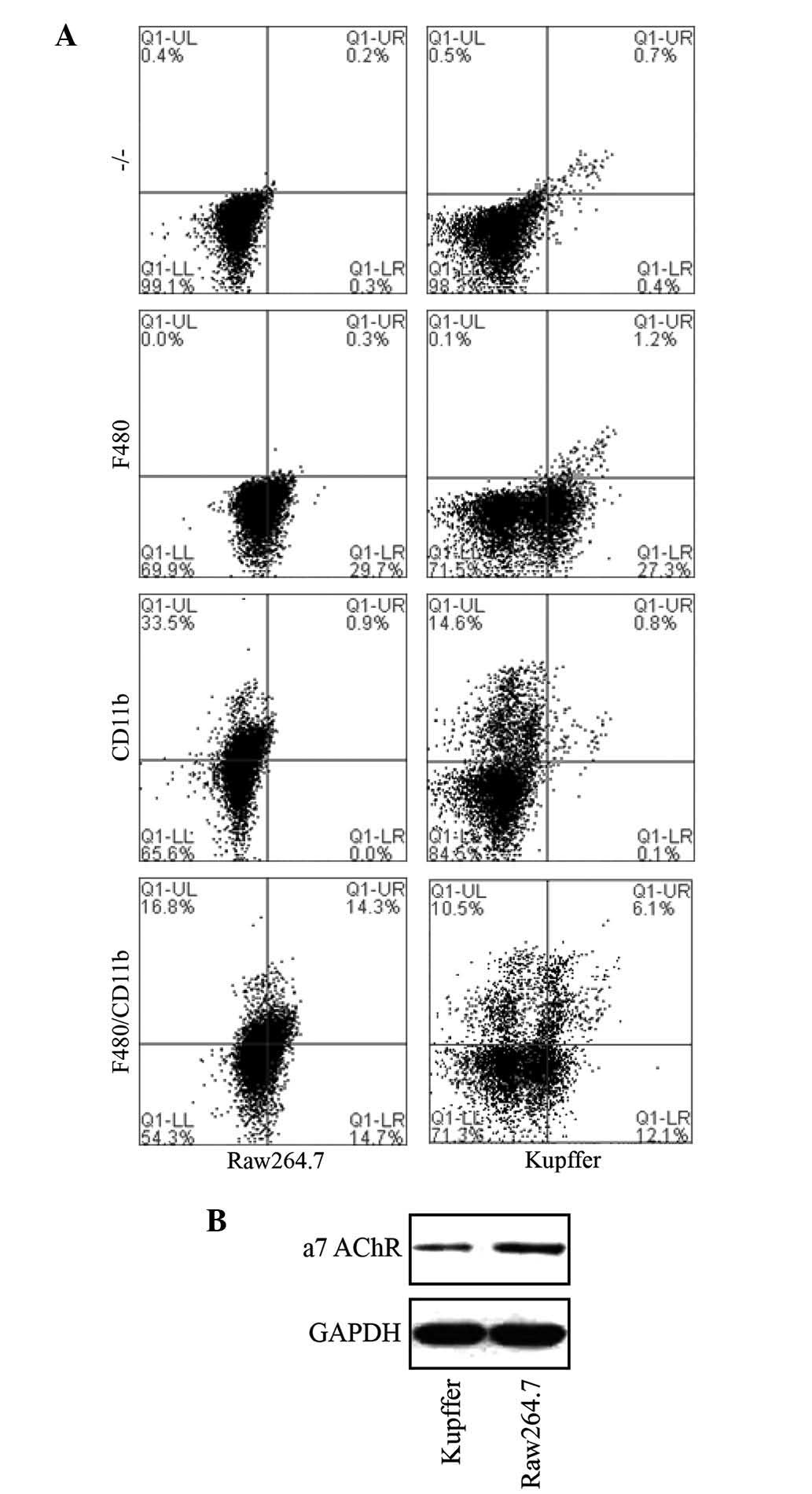
Nicotine exerts anti-inflammatory effects
via targeting a7 nAChR and inhibiting the NF-κB and ERK
pathways
To investigate the underlying mechanism of the
reduction of pro-inflammatory cytokines TNF-α and IL-6 by α7 nAChR
activation, primary macrophages from the liver were isolated and
assessed. Primary liver Kupffer cells were successfully isolated
and identified by staining with CD11b and F480 macrophage-specific
markers (Caltag Laboratories, Burlingame, CA, USA) and flow
cytometric detection (BD-Accuri C6, BD Biosciences Franklin Lakes,
NJ, USA) in comparison with murine macrophage RAW 264.7 cells
(Fig. 4).
In the mouse model of NASH, the α7 nAChR agonist
nicotine significantly attenuated HFD-induced upregulation of serum
TNF-α and IL-6. To determine the anti-inflammatory mechanisms of α7
nAChR-activation, the present study assessed whether nicotine
treatment blocked the production of TNF-α and IL-6 with or without
α7 nAChR antagonist α-bungarotoxin (α-BGT) in the primary liver
Kupffer cells. The secretion of the classic inflammatory factors
TNF-α and IL-6 was detected by ELISA. Stimulation with
lipopolysaccharide (LPS) significantly upregulated the secretion of
TNF-α (Fig. 5A) and IL-6 (Fig. 5B) in the cell culture supernatants.
However, nicotine treatment significantly attenuated LPS-induced
upregulation of TNF-α (Fig. 5A)
and IL-6 (Fig. 5B) in a
dose-dependent manner. Furthermore, α7 nAChR antagonist α-BGT
blocked the nicotine-induced reduction of TNF-α (Fig. 5A) and IL-6 (Fig. 5B), which indicated that nicotine
reduced the production of TNF-α and IL-6 via activating α7
nAChR.
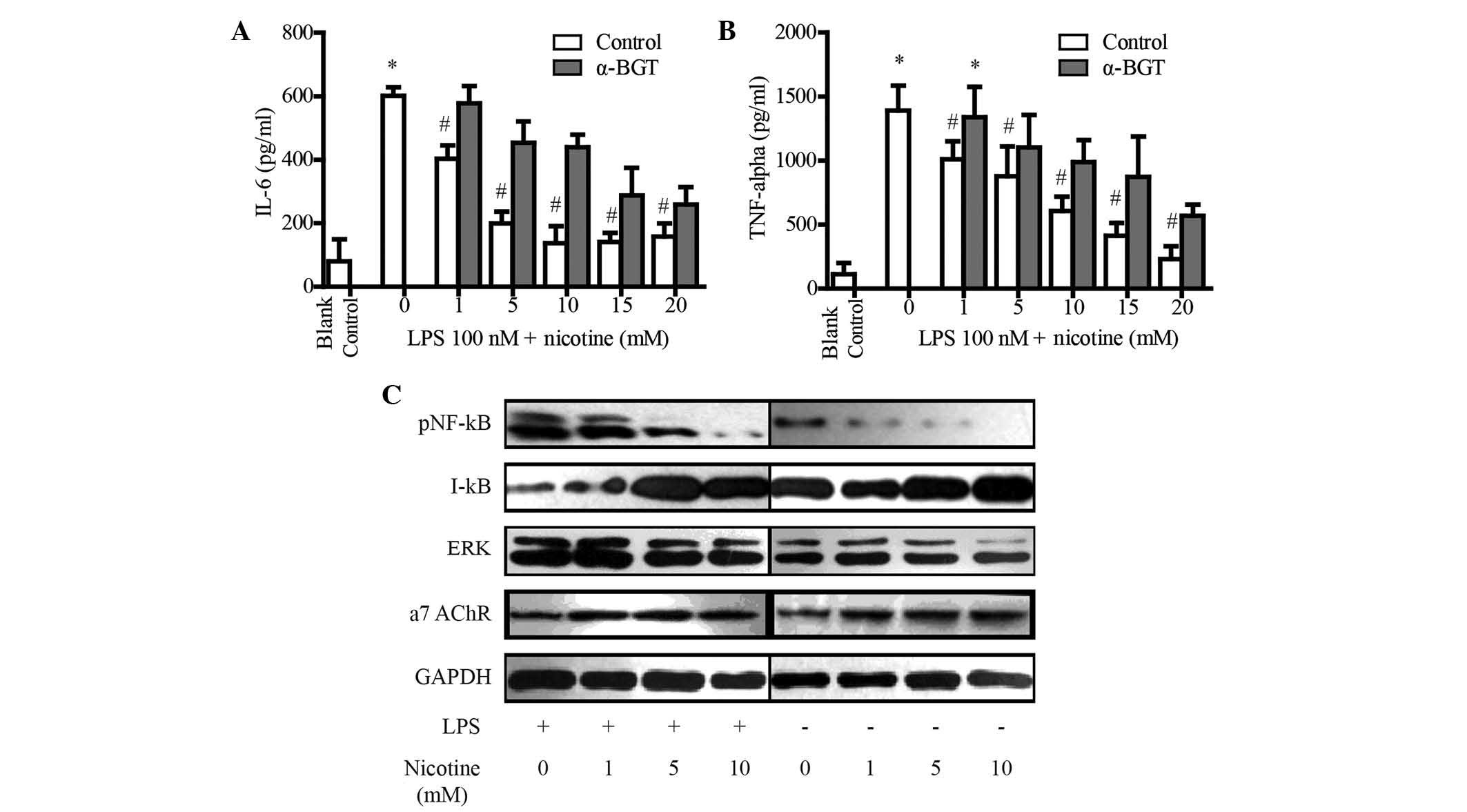 | Figure 5Nicotine reduces the secretion of
TNF-α and IL-6 in primary Kupffer cells via α7 nAChR. (A and B) The
supernatant was collected from the culture of Kupffer cells after
treatment with LPS and nicotine in the presence or absence of
α-BGT, and cytokine levels were assessed using ELISA kits. Values
are expressed as the mean ± standard deviation of three independent
experiments. *P<0.01 vs. blank control group;
#P<0.01 vs. LPS + 0 nM nicotine group. (C) Effects of
nicotine on the NF-κB and ERK pathway in primary Kupffer cells.
GAPDH was used as a loading control. Blots shown are representative
of at least three independent experiments. TNF, tumor necrosis
factor; IL, interleukin; NF-κB, nuclear factor kappa B; p,
phosphorylated, I-κB, inhibitor of NF-κB; ERK, extracellular
signal-regulated kinase; α7 nAChR, α7-nicotinic acetylcholine; LPS,
lipopolysaccharide; α-BGT, α7 nAChR antagonist α-bungarotoxin. |
Release of inflammatory cytokines is mostly mediated
via the ERK and NF-κB pathways, and ERK and NF-κB are the main
downstream signaling molecules of α7 nAChR (27). The present study therefore
investigated whether activation of α7 nAChR reduces the production
of cytokines via inhibiting the ERK and NF-κB pathways (Fig. 5C). As shown in Fig. 5, nicotine treatment upregulated the
protein levels of α7 nAChR in Kupffer cells in a dose-dependent
manner, which was consistent with the results of a previous study
(27). Furthermore, nicotine
obviously downregulated ERK and NF-κB levels in Kupffer cells.
Discussion
The results present study suggested that specific
interference with α7 nAChR represents a novel strategy for the
treatment of NASH. It was shown that treatment with the α7 nAChR
agonist nicotine for three weeks obviously attenuated hepatic
steatosis and reduced the production of TNF-α and IL-6 in an
HFD-induced mouse model of NASH. To investigate the underlying
mechanism, the primary macrophages from mouse livers were isolated
and treated with nicotine. The results showed that nicotine reduced
LPS-induced secretion of TNF-α and IL-6 in vitro, which was
blocked by α7 nAChR antagonist α-BGT. These results indicated that
nicotine suppressed TNF-α and IL-6 secretion by LPS-stimulated
macrophages through α7 nAChR activation. Furthermore, the present
study showed that nicotine-stimulated α7 nAChR activation
significantly downregulated NK-κB and ERK. It appeared that the
activation of α7 nAChR suppressed the production of
pro-inflammatory cytokines through NK-κB and ERK pathways.
NASH is increasingly recognized as a major
epidemiological problem, linking the metabolic syndrome to liver
fibrosis, cirrhosis and hepatocellular carcinoma. Currently
discussed treatment options comprise drugs approved for managing
the symptoms of impaired glucose metabolism, hypertension and
hyperlipidemia, including angiotensin I antagonists or insulin
sensitizers (30). However, the
incidence of NAFLD in the human population is further increasing,
affecting up to 30% of the general population worldwide, despite
the availability of these drugs (31). In addition, the side effects of
approved drugs preclude treatment of patient sub-populations, thus
underlining the requirement for additional specific treatment
options (27).
To test the effects of α7 nAChR activation on NASH,
the HFD-induced mouse model of NASH was employed. The model
developed symptoms within a time frame of 18 weeks and was
characterized by a rather mild elevation in liver enzymes, such as
ALT, in the circulation as well as the presence of lobular
inflammation, which is also observed in humans with NASH (32). The present study showed that the α7
nAChR agonist nicotine reduced NASH-associated hepatic steatosis in
mouse models. Furthermore, nicotine treatment decreased the
secretion of the pro-inflammatory cytokines TNF-α, IL-6 in mice
with NASH. This result indicated that activation of α7 nAChR and
the resulting anti-inflammatory effects may represent a novel
therapeutic strategy for NASH.
Inflammation characterized by the release of soluble
factors, including chemokines and cytokines, in addition to immune
cell activation, is regarded as an integral part of NASH and
several lines of evidence suggested that targeting of inflammation
is a promising tool for the management of NASH (29). The results of the present study
showed that a7 nAChR agonist nicotine reduced the production of
TNF-α and IL-6 in the mouse model of NASH in vivo and in
primary Kupffer cells in vitro. These results were
consistent with those of previous studies, which reported that
activation of the a7 nAChR expressed on resident macrophages may
suppress the local inflammation by reducing the production of
pro-inflammatory cytokines TNF-α and IL-6 (27). Furthermore, the present study found
that nicotine-induced a7 nAChR activation significantly inhibited
the expression of NF-κB and ERK. This result indicated that
activation of the a7 nAChR may inhibit cytokine production by
Kupffer cells via the NF-κB and ERK pathways.
In conclusion, the present study indicated that
modulating the inflammatory response in affected livers via
activating a7 nAChR may represent a novel strategy for the
treatment of NASH. The feasibility of this strategy requires
pre-clinical and clinical validation in further studies.
Acknowledgments
This work was supported by the National Natural
Science Foundation of China (grant no. 81100279) and the Foundation
of Zhejiang Health Committee (no. 2013KYA145).
References
|
1
|
Noureddin M, Yates KP, Vaughn IA,
Neuschwander-Tetri BA, Sanyal AJ, McCullough A, Merriman R, Hameed
B, Doo E, Kleiner DE, et al: Clinical and histological determinants
of nonalcoholic steatohepatitis and advanced fibrosis in elderly
patients. Hepatology. 58:1644–1654. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Evans CD, Oien KA, MacSween RN and Mills
PR: Non-alcoholic steatohepatitis: A common cause of progressive
chronic liver injury? J Clin Pathol. 55:689–692. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ascha MS, Hanouneh IA, Lopez R, Tamimi TA,
Feldstein AF and Zein NN: The incidence and risk factors of
hepatocellular carcinoma in patients with nonalcoholic
steatohepatitis. Hepatology. 51:1972–1978. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Adams LA, Lymp JF, St Sauver J, Sanderson
SO, Lindor KD, Feldstein A and Angulo P: The natural history of
nonalcoholic fatty liver disease: A population-based cohort study.
Gastroenterology. 129:113–121. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dunn W, Xu R, Wingard DL, Rogers C, Angulo
P, Younossi ZM and Schwimmer JB: Suspected nonalcoholic fatty liver
disease and mortality risk in a population-based cohort study. AM J
Gastroenterol. 103:2263–2271. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wong VW, Wong GL, Tsang SW, Fan T, Chu WC,
Woo J, Chan AW, Choi PC, Chim AM, Lau JY, et al: High prevalence of
colorectal neoplasm in patients with non-alcoholic steatohepatitis.
Gut. 60:829–836. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gastaldelli A, Kozakova M, Højlund K,
Flyvbjerg A, Favuzzi A, Mitrakou A and Balkau B: Fatty liver is
associated with insulin resistance, risk of coronary heart disease
and early atherosclerosis in a large European population.
Hepatology. 49:1537–1544. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bilici A, Ozguroglu M, Mihmanli I, Turna H
and Adaletli I: A case-control study of non-alcoholic fatty liver
disease in breast cancer. Med Oncol. 24:367–371. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pagadala MR, Zein CO, Dasarathy S, Yerian
LM, Lopez R and McCullough AJ: Prevalence of hypothyroidism in
nonalcoholic fatty liver disease. Dig Dis Sci. 57:528–534. 2012.
View Article : Google Scholar
|
|
10
|
Caballería L, Auladell MA, Torán P,
Miranda D, Aznar J, Pera G, Gil D, Muñoz L, Planas J, Canut S, et
al: Prevalence and factors associated with the presence of non
alcoholic fatty liver disease in an apparently healthy adult
population in primary care units. BMC Gastroenterol. 7:412007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gelpi Méndez JA, Castellanos Fillot A,
Sainz Gutiérrez JC, Quevedo Aguado L and Martin Barallat J:
Prevalence of non-alcoholic fatty liver disease and associated risk
factors among managers from the community of Madrid. Arch Prev
Riesgos Labor. 17:84–90. 2014.In Spanish. View Article : Google Scholar
|
|
12
|
Leite NC, Salles GF, Araujo AL,
Villela-Nogueira CA and Cardoso CR: Prevalence and associated
factors of non-alcoholic fatty liver disease in patients with
type-2 diabetes mellitus. Liver Int. 29:113–119. 2009. View Article : Google Scholar
|
|
13
|
Loria P, Lonardo A, Lombardini S, Carulli
L, Verrone A, Ganazzi D, Rudilosso A, D'Amico R, Bertolotti M and
Carulli N: Gallstone disease in non-alcoholic fatty liver:
Prevalence and associated factors. J Gastroen Hepatol.
20:1176–1184. 2005. View Article : Google Scholar
|
|
14
|
Radu C, Grigorescu M, Crisan D, Lupsor M,
Constantin D and Dina L: Prevalence and associated risk factors of
non-alcoholic fatty liver disease in hospitalized patients. J
Gastrointestin Liver Dis. 17:255–260. 2008.PubMed/NCBI
|
|
15
|
Hu KC, Wang HY, Liu SC, Liu CC, Hung CL,
Bair MJ, Liu CJ, Wu MS and Shih SC: Nonalcoholic fatty liver
disease: Updates in noninvasive diagnosis and correlation with
cardiovascular disease. World J Gastroenterol. 20:7718–7729. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sanyal AJ: NASH: A global health problem.
Hepatol Res. 41:670–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Takaki A, Kawai D and Yamamoto K:
Molecular mechanisms and new treatment strategies for non-alcoholic
steatohepatitis (NASH). Int J Mol Sci. 15:7352–7379. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Musso G, Anty R and Petta S: Antioxidant
therapy and drugs interfering with lipid metabolism: could they be
effective in NAFLD patients? Curr Pharm Des. 19:5297–5313. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yalcin M, Akarsu M, Celik A, Sagol O,
Tunali S, Ertener O, Bengi G and Akpinar H: A comparison of the
effects of infliximab, adalimumab, and pentoxifylline on rats with
non-alcoholic steatohepatitis. Turk J Gastroenterol. 25:167–175.
2014. View Article : Google Scholar
|
|
20
|
Fock KM and Khoo J: Diet and exercise in
management of obesity and overweight. J Gastroenterol Hepatol.
28:59–63. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Beaton MD: Current treatment options for
nonalcoholic fatty liver disease and nonalcoholic steatohepatitis.
Can J Gastroenterol. 26:353–357. 2012.PubMed/NCBI
|
|
22
|
Malaguarnera M, Di Rosa M, Nicoletti F and
Malaguarnera L: Molecular mechanisms involved in NAFLD progression.
J Mol Med (Berl). 87:679–695. 2009. View Article : Google Scholar
|
|
23
|
Satapathy SK, Garg S, Chauhan R, Sakhuja
P, Malhotra V, Sharma BC and Sarin SK: Beneficial effects of tumor
necrosis factor-alpha inhibition by pentoxifylline on clinical,
biochemical and metabolic parameters of patients with nonalcoholic
steatohepatitis. AM J Gastroenterol. 99:1946–1952. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mitchel EB and Lavine JE: Review article:
the management of paediatric nonalcoholic fatty liver disease.
Aliment Pharmacol Ther. 40:1155–1170. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Filippini P, Cesario A, Fini M, Locatelli
F and Rutella S: The Yin and Yang of non-neuronal α7 -nicotinic
receptors in inflammation and autoimmunity. Curr drug targets.
13:644–655. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Boeckxstaens G: The clinical importance of
the anti-inflammatory vagovagal reflex. Handb Clin Neurol.
117:119–134. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ganz M and Szabo G: Immune and
inflammatory pathways in NASH. Hepatol int. 7:771–781. 2013.
View Article : Google Scholar
|
|
28
|
Meli R, Mattace Raso G and Calignano A:
Role of innate immune response in non-alcoholic Fatty liver
disease: metabolic complications and therapeutic tools. Front
Immunol. 5:1772014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Braunersreuther V, Viviani GL, Mach F and
Montecucco F: Role of cytokines and chemokines in non-alcoholic
fatty liver disease. World J Gastroenterol. 18:727–735. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Weiß J, Rau M and Geier A: Non-alcoholic
fatty liver disease epidemiology, clinical course, investigation,
and treatment. Dtsch Arztebl Int. 111:447–452. 2014.
|
|
31
|
Ratziu V, Goodman Z and Sanyal A: Current
efforts and trends in the treatment of NASH. J Hepatol. 62:S65–S75.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gadd VL, Skoien R, Powell EE, Fagan KJ,
Winterford C, Horsfall L, Irvine K and Clouston AD: The portal
inflammatory infiltrate and ductular reaction in human nonalcoholic
fatty liver. Hepatology. 59:1393–1405. 2014. View Article : Google Scholar
|