Introduction
Non-alcoholic steatohepatitis (NASH) is
characterized by hepatic fat accumulation, inflammation and varying
degrees of fibrosis. The central pathophysiological issue in
patients afflicted with NASH is insulin resistance. Improvement of
insulin resistance has therapeutic potential in preventing the
progression of NASH (1).
Previously, dipeptidyl peptidase (DPP)-IV inhibitors have been used
as novel oral drugs for the treatment of type 2 diabetes. DPP-IV is
an enzyme, which inactivates incretins, including glucagon-like
peptide-1 and gastric inhibitory polypeptide, which regulates blood
glucose primarily via stimulation of glucose-dependent insulin
release. As a result, the activation of DPP-IV leads to the
development of glucose intolerance and hepatic steatosis (2). DPP-IV enzyme has widespread organ
distribution throughout the body and has pleiotropic biological
functions (3–5). DPP-IV is involved in glucose
metabolism and lipid accumulation, degradation of the extracellular
matrix, appetite regulation and immune stimulation via its
peptidase activities (6–10). The liver is one of the organs
expressing DPP-IV to a high degree (11). The mRNA expression of DPP-IV is
also increased in the liver of non-alcoholic fatty liver disease
(NAFLD) (12). Serum DPP-IV
activity and the expression of hepatic DPP-IV are correlated with
hepatic steatosis and NAFLD severity (13). DPP-IV deficient rats exhibit a
reduction of hepatic steatosis, hepatic inflammatory and
pro-fibrogenic cytokines compared with the wild-type rats (14). Therefore, DPP-IV may be involved in
not only insulin resistance, but also hepatic lipogenesis,
inflammation and fibrosis during the progression of NASH.
Therefore, DPP-IV inhibitors may have favorable effects for four
pathways in the treatment of NASH, including insulin resistance,
hepatic lipogenesis, inflammation and fibrogenesis.
Fatty liver Shionogi (FLS)-ob/ob mice
are generated by transferring the Lepob gene into
the genome of FLS mice without obesity, which then spontaneously
develop chronic hepatic steatosis. The features of
FLS-ob/ob mice are hyperphagia, obesity,
hyperlipidemia and diabetes mellitus (15). They exhibit histologically severe
steatosis, hepatocellular ballooning and advanced hepatic fibrosis,
increased oxidative stress, elevated inflammatory, as well as
pro-fibrotic cytokine production, increased apoptosis of
hepatocytes and the mice also develop cirrhosis and liver tumors
(16,17). FLS-ob/ob mice are the
closest animal model to metabolic syndrome-associated NASH in
humans and possess targets of DPP-IV inhibitors. Previously,
sitagliptin, a DPP-IV inhibitor, was reported to attenuate
methionine/choline-deficient (MCD) diet-induced steatohepatitis
(18). However, this model is
markedly different compared with the features of human NASH. The
present study aimed to confirm whether sitagliptin improved
steatohepatitis in the FLS-ob/ob mice by modifying
insulin resistance, hepatic lipogenesis, inflammation, fibrosis and
oxidative stress.
Materials and methods
Animals
Twenty male FLS-ob/ob mice (age, 8
weeks; body weight, 40.14±3.39 g) were obtained from Shionogi
Research Laboratories (Shiga, Japan) and housed under a controlled
temperature of 24±2°C and a 12-h light/dark cycle. The mice were
provided with water and standard powder chow (CE-2, 4.6% fat; CLEA
Japan, Inc., Tokyo, Japan) ad libitum. Food consumption and
body weight were monitored throughout the observation period to
equalize the dietary intake between the two groups. All experiments
were performed in accordance with the Animal Experimentation
Guidelines of Tottori University (Yonago, Japan). The study was
approved by the ethics committee of Tottori University, Yonago,
Japan (approval no. 11-Y-54).
Administration of sitagliptin
Male FLS-ob/ob 12-week-old mice were
randomly assigned to the control or sitagliptin group (n=10/group).
Sitagliptin (2 mg/kg/day; Merck & Co., Inc, Kenilworth, NJ,
USA) was administered as a bolus every afternoon per os. for
12 weeks through a gastric tube. The control group was administered
water. Blood was drawn from the tail vein after 4 h fasting and the
blood glucose level was measured every 4 weeks. After 12 weeks, the
animals were sacrificed under pentobarbital anesthesia (Dainippon
Sumitomo Pharma, Osaka, Japan) and blood was collected from the
right ventricle. The plasma samples were frozen and stored at
−80°C. The liver and visceral fat were weighed, snap frozen in
liquid nitrogen and stored at −80°C. Liver specimens were also
fixed in 10% buffered formalin (Wako Pure Chemical Industries,
Ltd., Osaka, Japan) and embedded in paraffin (Wako Pure Chemical
Industries, Ltd.) for histological analysis.
Analysis of hepatic cholesterol and
triglyceride contents
Snap frozen liver samples (50 mg) were homogenized
and extracted with chloroform-methanol (2:1, v/v; Wako Pure
Chemical Industries, Ltd.), and subsequently the organic phase was
dried and resuspended in 2-propanol, containing 10% Triton X-100.
The total cholesterol and triglyceride levels were measured using a
Cholesterol E-test (Wako Pure Chemical Industries, Ltd.) and a
Triglyceride E-test (Wako Pure Chemical Industries, Ltd.),
respectively.
Biochemical analyses
The blood samples were immediately separated via
centrifugation at 2,000 × g for 15 min at 4°C, and were stored at
−80°C until further use. The serum samples were analyzed to
determine the levels of aspartate aminotransferase (AST) and
alanine aminotransferase (ALT).
Measurement of areas of hepatic
steatosis
Neutral lipids in frozen-fixed, cryostat-embedded
liver sections (4-µm thick) were stained with Oil Red O
(Sigma-Aldrich, St. Louis, MO, USA). The areas of hepatic steatosis
were subsequently measured in 10 randomly selected fields
(magnification, ×400; Olympus BX51N-34; Olympus Corporation, Tokyo,
Japan) in each specimen using Win ROOF version 5.71 software
(Mitani Corporation, Tokyo, Japan).
Measurement of hepatic fibrosis area with
sirius red staining
Formalin-fixed, paraffin-embedded liver sections (4
µm thick) were stained with Picrosirius red
(Chroma-Gesellschaft Schmid GmbH & Co., Münster, Germany) and
counterstained with fast green (Chroma-Gesellschaft Schmid GmbH
& Co.). The areas of hepatic fibrosis were subsequently
measured in 10 randomly selected fields in each specimen
(magnification, ×400) using Win ROOF version 5.71 software and the
Olympus BX51N-34 microscope.
Immunostaining for α-smooth muscle actin
(SMA)
The present study immunohistochemically detected
α-SMA by staining with mouse monoclonal anti-α-SMA antibody (cat.
no. MS-113-R7; Thermo Fisher Scientific, Fremont, CA, USA) without
dilution. Goat anti-mouse Ig, from the Histofine® Mouse
Stain kit (cat. no. 414322; Nichirei Biosciences, Inc., Tokyo,
Japan), was used as the secondary antibody without dilution. The
activation of hepatic stellate cells (HSC) was assessed by
measuring the areas of α-SMA staining using Win ROOF version 5.71
software in 10 randomly selected fields (magnification, ×400;
Olympus BX51N-34 microscope) in each specimen.
Analysis of inflammatory cell
infiltration of liver tissue
F4/80, which is a mature mouse cell surface
glycoprotein expressed at high levels on Kupffer cells (19), was immunohistochemically stained
using a rat monoclonal anti-F4/80 mouse antibody (cat. no. ab6640;
Abcam, Tokyo, Japan) diluted at 1:100 with 0.01 M/l
phosphate-buffered saline (PBS), according to the manufacturer's
instructions. Goat anti-rat secondary antibody, from the
Histofine® Simple Stain™ Mouse MAX-PO (Rat) kit (cat.
no. 414311; Nichirei Biosciences, Inc.) was used without dilution.
The immunopositive cells were analyzed in 10 intralobular ocular
fields (magnification, ×400; Olympus BX51N-34 microscope) in each
specimen.
Analysis of oxidative stress
Oxidative stress was assessed by immunohistochemical
staining to detect 8-hydroxy-2-deoxyguanosine (8-OHdG), a marker of
oxidative DNA damage (16), using
a monoclonal mouse anti-8-OHdG antibody (cat. no. MOG-020P; Nikken
SEIL, Shizuoka, Japan), diluted with 200 µl distilled water,
according to the manufacturer's instructions. Goat anti-mouse Ig,
from the Histofine® Mouse Stain kit, served as the
secondary antibody without dilution. The immunopositive cells were
analyzed using Win ROOF version 5.71 software in 10 intralobular
ocular fields (magnification, ×400; Olympus BX51N-34 microscope) in
each specimen, and the values were expressed as the ratios (%) of
fields. Furthermore, the present study semi-quantified
4-hydroxynonenal (4-HNE), which was immunohistochemically stained
using a monoclonal mouse anti-4-HNE antibody (cat. no. MHN-020P;
Nikken SEIL), diluted with 200 µl distilled water, according
to the manufacturer's instructions. Goat anti-mouse Ig, from the
Histofine® Mouse Stain kit, was used as the secondary
antibody without dilution. A total of 10 randomly selected fields
in each specimen, which were stained with 4-HNE (magnification,
×400) were classified into immunopositive grades 1, 2, 3 or 4
(0–10%, 11–20%, 21–30% and >30%, respectively) and the mean
values of the 10 fields were calculated.
Analysis of apoptotic cells in liver
tissue
The apoptotic cells in liver tissue were detected
in situ by specific labeling of nuclear DNA fragmentation
using terminal deoxynucleotidyl transferase dUTP nick end labeling
(TUNEL). The sections were deparaffinized in xylene, rehydrated,
washed with PBS and digested with 20 µg/ml proteinase K
(Wako Pure Chemical Industries, Ltd.) for 10 min at room
temperature. The fragmented DNA was detected via the TUNEL method
using the Apop Tag Plus Peroxidase In Situ Apoptosis
Detection kit (EMD Millipore, Billerica, MA, USA), according to the
manufacturer's instructions. The numbers of stained and unstained
cells were counted using Win ROOF version 5.71 software in 10
intralobular ocular fields (magnification, ×400) in each
specimen.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analysis
Hepatic tissue samples were homogenized and the
total RNA was extracted using the RNeasy Lipid Tissue Mini kit
(Qiagen, Hilden, Germany). The RNA concentrations were determined
by measuring the absorbance at 260 nm using a NanoDrop 1000
spectrophotometer (Thermo Fisher Scientific), and the RNA quality
was confirmed by electrophoresis on ethidium bromide stained 1%
agarose gels. The total RNA (2 µg) was reverse transcribed
in a final volume of 11.5 µl, containing 4 µl 5X
standard buffer, 2 µl 0.1 M dithiothreitol, 1 µl
SuperScript II RNase H reverse transcriptase (Invitrogen Life
Technologies, Carlsbad, CA, USA), 2 µl 10 mM dNTP (Promega,
Madison, WI, USA), 1 µl 50 pmol/µl random primer
(Promega), 0.5 µl 100 pmol/µl oligo (dt) 15 Primer
(Promega) and 1 µl 40 U/µl ribonuclease inhibitor
(Wako Pure Chemical Industries, Ltd.). The mixtures were incubated
at 37°C for 60 min, 95°C for 5 min and subsequently cooled to 4°C
for 5 min using a MyCycler™ Thermal Cycler (Bio-Rad Laboratories,
Inc., Hercules, CA, USA).
Real-time PCR
Quantitative real-time PCR assays (7900HT Fast
Realtime PCR system; Applied Biosystems Life Technologies, Foster
City, CA, USA) were performed in a final volume of 10 ml,
containing 250 nM Universal ProbeLibrary probe (Roche, Basel,
Switzerland), 900 nM forward primer, 900 nM reverse primer, 5 ml
EXPRESS qPCR Supermix with Premixed Rox (Invitrogen Life
Technologies) and 2 ml cDNA. The mRNA expression levels of
transforming growth factor-β1 (TGF-β1; GenBank, NM_011577),
procollagen-type I (GenBank, U08020), connective tissue growth
factor (CTGF; GenBank, NM_010217), tumor necrosis factor-α (TNF-α;
GenBank, NM_013693), monocyte chemoattractant protein-1 (MCP-1;
GenBank, NM_100127112), tissue inhibitor of metalloproteinases-1
(TIMP-1; GenBank, NM_011593), peroxisome proliferator activated
receptor (PPAR-α; GenBank, NM_007988.3), sterol regulatory
element-binding protein 1c (SREBP1c; GenBank, NM_011480), fatty
acid synthase (FAS; GenBank, AF127033) and microsomal triglyceride
transfer protein (MTP; GenBank, NM_008642) were assessed using the
7900HT Fast Real-Time PCR system with SDS2.3 software (Applied
Biosystems Life Technologies) and β-actin (GenBank, NM_007393) was
used as an internal standard. The thermal cycle conditions were as
follows: 95°C for 20 sec, followed by 45 cycles of 1 sec at 95°C
and 20 sec at 60°C. The relative mRNA expression levels were
calculated using the 2−ΔΔCT method (20).
Statistical analysis
The significance of the differences between the
groups was statistically analyzed using an unpaired Student's
t-test. The data were statistically analyzed using StatFlex version
6.0 for Windows software (Artech Co, Ltd., Osaka, Japan). All data
are expressed as the mean ± standard deviation. P<0.05 was
considered to indicate a statistically significant difference.
Results
Characteristics of the mice
As shown in Table
I, the body weight of the mice was significantly lower in the
sitagliptin group compared with the control group. The liver weight
and liver-to-body weight ratio in the sitagliptin group also
demonstrated a decreased tendency compared with the control group.
Food consumption and visceral fat weight revealed no significant
difference between the two groups. The serum levels of AST and ALT
also revealed no significant difference between the two groups. The
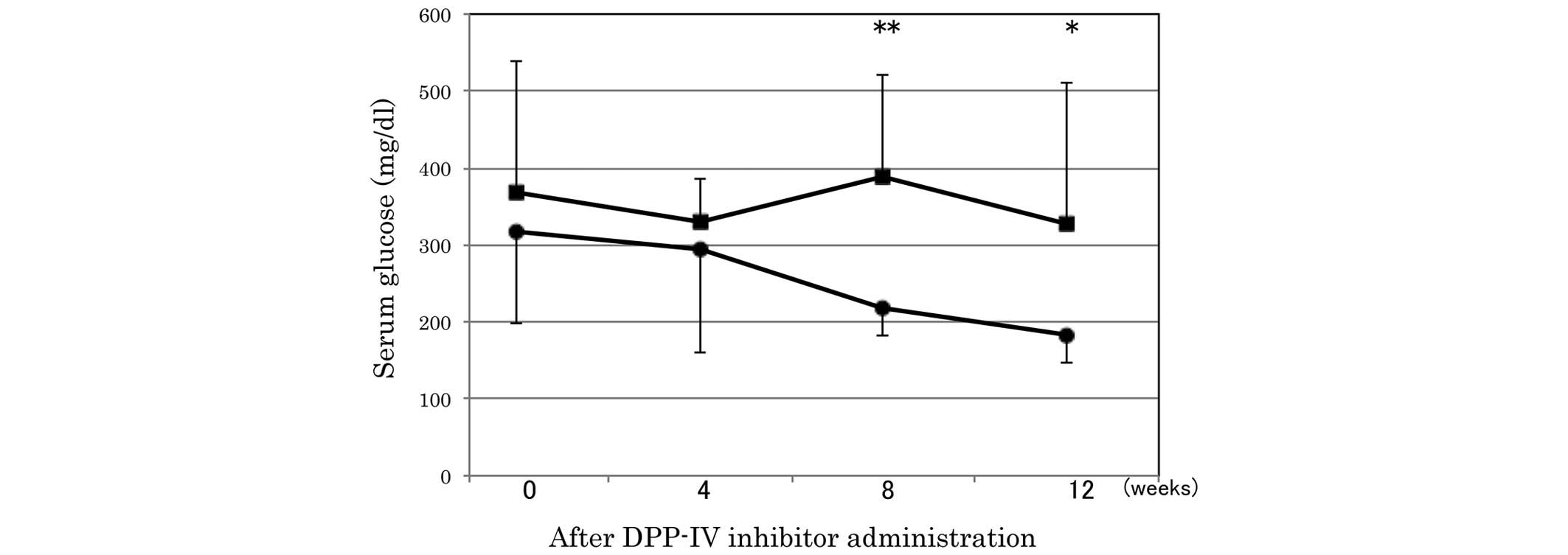
serum glucose was significantly lower in the sitagliptin group
compared with the control group at 8 and 12 weeks after the
administration of the DPP-IV inhibitor (Fig. 1).
 | Table IEffects of sitagliptin administration
on various characteristics of the mice. |
Table I
Effects of sitagliptin administration
on various characteristics of the mice.
| Characteristic | Control group
(n=10) | Sitagliptin group
(n=10) |
|---|
| Body weight
(g) | 56±8 | 48±9a |
| Liver weight
(g) | 7.8±2.5 | 6.0±2.7 |
| Liver/body weight
ratio | 0.14±0.03 | 0.12±0.04 |
| Visceral fat weight
(g) | 2.3±0.4 | 2.2±0.6 |
| Weekly dietary
intake (g) | 37±12 | 36±10 |
| Serum AST
(U/l) | 183±73 | 158±61 |
| Serum ALT
(U/l) | 226±107 | 232±195 |
Effects of sitagliptin on hepatic
steatosis
To assess the effects of sitagliptin on lipid
metabolism, the present study determined the hepatic steatosis
area, hepatic lipid contents and gene expression of hepatic
lipogenesis, lipolysis and lipid transporter. Oil Red O staining
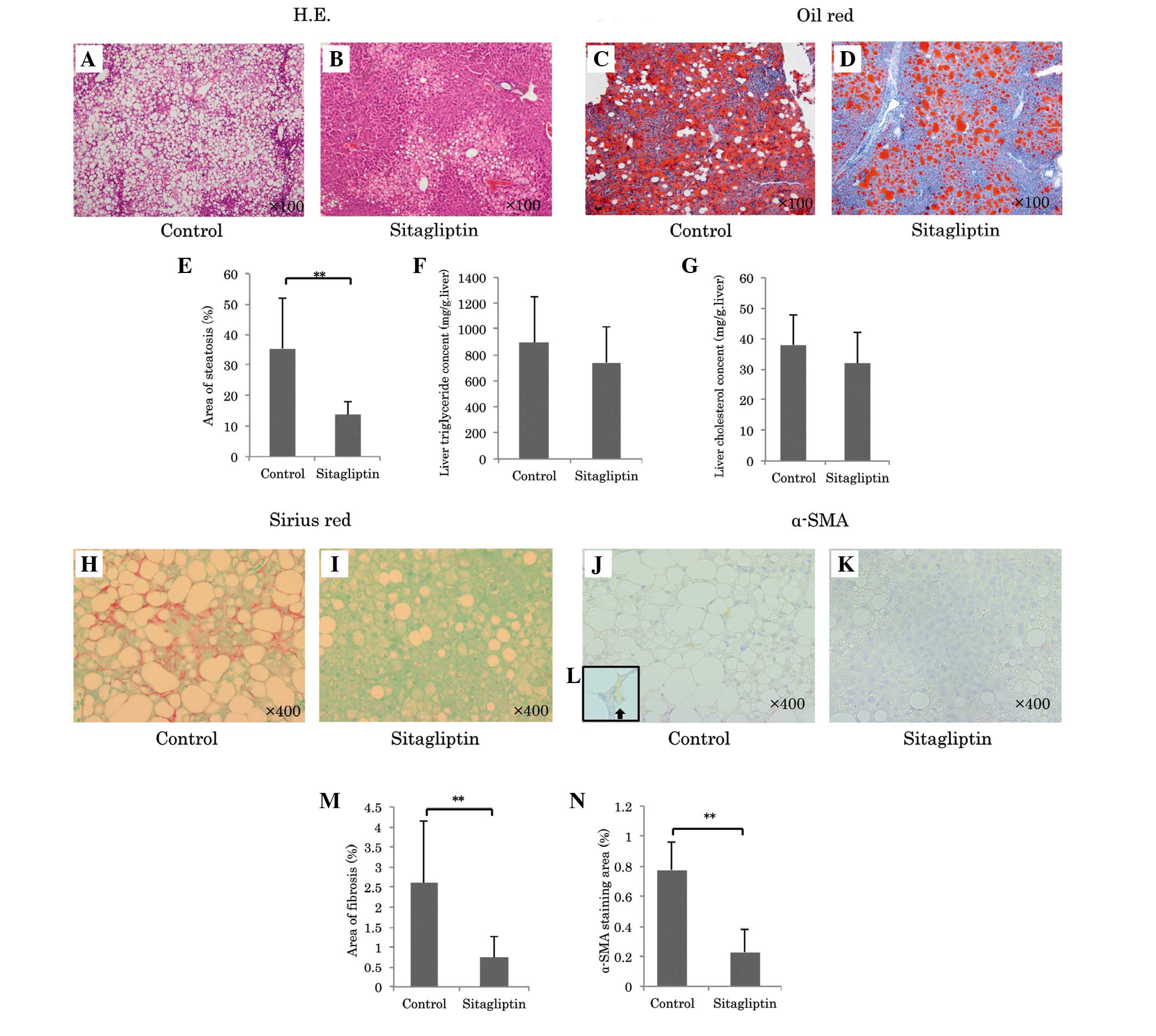
revealed that sitagliptin significantly reduced the area of hepatic
steatosis (sitagliptin group, vs. control group; 13.8±4.3, vs.
35.4±16.5%; P<0.001; Fig.
2A-E). Hepatic total cholesterol and triglyceride contents in
the sitagliptin group were lower compared with the control group
(Hepatic cholesterol: Sitagliptin, 31.8±10.3, vs. control, 37.9±9.9
mg/g liver; Triglyceride: Sitagliptin, 741±276, vs. control:
894±359 mg/g liver), however, not significantly so (Fig. 2F and G). The mRNA expression levels
of genes associated with hepatic steatosis were listed in Table II. The mRNA expression of PPAR-α
was significantly increased in the sitagliptin group (sitagliptin,
vs. control group; 2.83±0.59, vs. 2.07±0.63; P=0.0095). The mRNA
expression of FAS was significantly decreased in the sitagliptin
group (sitagliptin, vs. control group; 1.65±0.43, vs. 6.21±3.78;
P=0.0013). The mRNA expression levels of SREBP1c and MTP were
reduced in the sitagliptin group compared with the control,
however, not significantly so. Taken together, these findings
suggested that sitagliptin reduced lipid synthesis and the
accumulation in the liver of FLS-ob/ob mice.
| Table IImRNA expression levels of various
genes in the control and sitagliptin groups. |
Table II
mRNA expression levels of various
genes in the control and sitagliptin groups.
| mRNA | Control group
(n=10) | Sitagliptin group
(n=10) |
|---|
| FAS | 6.21±3.79 | 1.65±0.43b |
| SREBP1c | 1.98±0.62 | 1.59±0.42 |
| PPAR-α | 2.07±0.63 | 2.83±0.59b |
| MTP | 1.63±0.37 | 1.44±0.28 |
| Procollagen-type
I | 24.89±17.47 | 8.06±6.40b |
| TGF-β1 | 2.40±0.46 | 1.65±0.47b |
| CTGF | 7.32±10.93 | 3.36±0.91 |
| TIMP-1 | 11.43±6.53 | 4.05±2.69b |
| TNF-α | 4.81±2.19 | 2.64±0.91b |
| MCP-1 | 8.10±5.10 | 4.30±2.45a |
Effects of sitagliptin on hepatic
fibrosis
To assess the possibility that sitagliptin reduced
hepatic fibrosis, the present study determined the antifibrotic
effects of sitagliptin in the FLS-ob/ob mice using
Sirius red staining, α-SMA staining and pro-fibrogenic cytokine
gene expression. Sirius red staining revealed that sitagliptin
reduced the area of fibrosis compared with the control
(sitagliptin, vs. control; 0.74±0.54, vs. 2.61±1.53%; P=0.0016;
Fig. 2H, I and M). Since activated
HSCs are a major contributor to hepatic fibrogenesis, the present
study measured the protein expression of α-SMA, which is expressed
by HSCs in response to liver injury. Notably, sitagliptin caused a
profound decrease in the area of positive α-SMA immunostaining
compared with the control (sitagliptin, vs. control; 0.23±0.15, vs.
0.78±0.19%; P<0.001; Fig. 2J-L and
N), which suggested that this treatment inhibited the
activation of HSCs. The mRNA expression of parameters associated
with extracellular matrix metabolism in the liver are listed in
Table II. Sitagliptin
significantly reduced the mRNA expression levels of procollagen I,
TGF-β1 and TIMP-1 compared with the controls. The mRNA expression
of CTGF tended to be lower in the sitagliptin group compared with
the control group.
Effects of sitagliptin on inflammatory
reactions in the liver
The process of hepatic fibrosis is driven primarily
by inflammation in response to liver damage. Sitagliptin markedly
reduced the number of F4/80 positive cells, representing liver
macrophage Kupffer cells, compared with the control group
(sitagliptin, vs. control; 0.95±0.33, vs. 4.31±2.78; P=0.0013;
Fig. 3A-C and G) and reduced the
quantity of mRNA for MCP-1 by 53% and TNF-α by 55% (Table II).
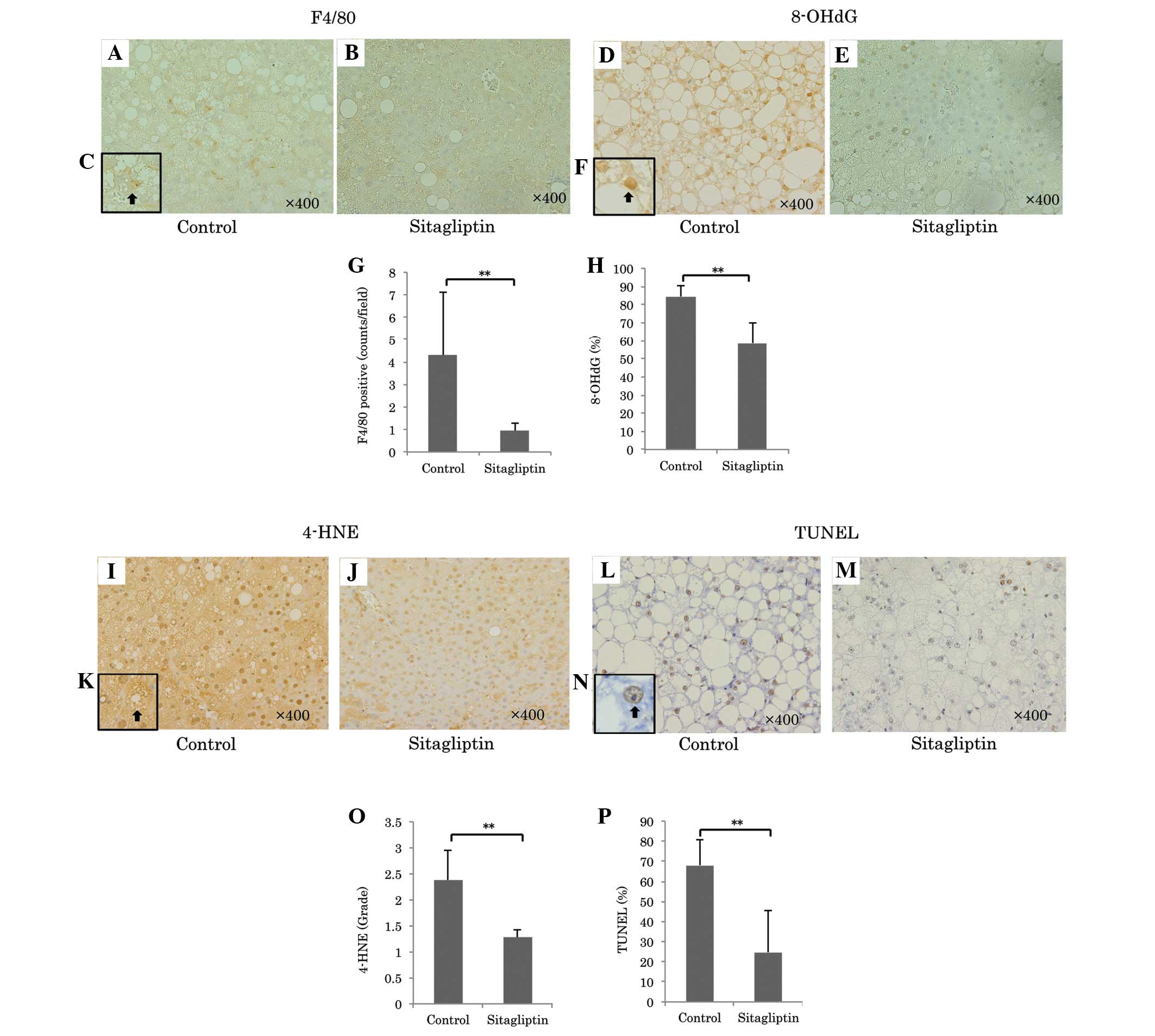 | Figure 3Representative images of F4/80
immunostaining for Kupffer cells (magnification, ×400) in the (A)
control and (B) sitagliptin groups. (C) Increased magnification
(×1,000) of immunopositive F4/80 positive cells. Representative
immunostaining for 8-OHdG (magnification, ×400) of the (D) control
and (E) sitagliptin groups. (F) A higher magnification of an 8-OHdG
positive nucleus (×1,000, arrow). (G) Quantitation of F4/80
immunopositive cells in each group indicated that F4/80
immunopositive cells were significantly decreased in the
sitagliptin group compared with the control
(**P<0.01). Oxidative stress was determined by 8-OHdG
and 4-HNE immunostaining. (H) A comparison of 8-OHdG immunopositive
cells between the groups (**P<0.01). Immunostaining
for 8-OHdG was significantly decreased in the sitagliptin group
compared with the control. Immunostaining for 4-HNE (magnification,
×400) in the (I) control and (J) sitagliptin groups. (K) 4-HNE
immunopositive cell at a higher magnification (×1,000, arrow).
Apoptotic cells were determined by TUNEL immunostaining
(magnification, ×400) in the (L) control and (M) sitagliptin
groups. (N) A higher magnification (×1,000) of a TUNEL positive
nucleus (arrow). (O) A comparison of 4-HNE immunopositive cells
between the groups. Immunostaining of 4-HNE was significantly
reduced in the sitagliptin group compared with the control
(**P<0.01). (P) A comparison of the numbers of TUNEL
positive cells between groups. The number of TUNEL-positive cells
was significantly decreased in the sitagliptin group compared with
the control (**P<0.01). 8-OHdG,
8-hydroxy-2-deoxyguanosine; 4-HNE, 4-hydroxynonenal; TUNEL,
terminal deoxynucleotidyl transferase dUTP nick end labeling. |
Effects of sitagliptin on oxidative
stress
Oxidative stress is involved in the development of
NASH. The present study determined oxidative stress by two methods:
8-OHdG as an index of DNA damage and 4-HNE as an index of a lipid
peroxidation. Sitagliptin markedly reduced the ratio of 8-OHdG
positive cells in the liver samples compared with the control
(sitagliptin, vs. control; 58.6±11.6, vs. 84.5±5.8%; P<0.001;
Fig. 3D-F and H) and significantly
reduced the immunostaining grade for liver 4-HNE (sitagliptin, vs.
control; 1.27±0.16, vs. 2.38±0.56; P<0.001; Fig. 3I-G and N).
Effect of sitagliptin on hepatic
apoptosis
Hepatocytes damaged by oxidative stress undergo
apoptosis. Sitagliptin significantly reduced the ratio of TUNEL
positive cells in the liver samples compared with the control
(sitagliptin, vs. control; 68.0±12.9, vs. 24.8±20.6%; P<0.001;
Fig. 3K-M and O).
Discussion
The present study revealed that sitagliptin
decreased blood glucose level, hepatic lipogenesis,
pro-inflammatory cytokines, pro-fibrogenic cytokines and oxidative
stress, consequently improving hepatic steatosis, inflammation and
fibrosis. The 'two-hit' theory and 'multiple parallel hits'
hypothesis have been proposed for the pathophysiology of NAFLD and
NASH (21). The first hit is
insulin resistance and leads to NAFLD. The following hits,
oxidative stress, cytokine production and inflammation increase,
result in the development of NASH (22–24).
Sitagliptin significantly improved hyperglycemia and
reduced body weight compared with the controls under the identical
dietary intake. The inhibition of DPP-IV by sitagliptin led to the
attenuation of incretin degradation, consequently increasing
insulin and reducing blood glucose levels. TNF-α leads to insulin
resistance by impairing insulin signaling and reducing glucose
transporters (22). The reduction
of the gene expression of TNF-α in the sitagliptin group also
improved insulin resistance. The loss of body weight by DPP-IV
inhibitors is suggested to be due to two mechanisms, one of which
is the loss of appetite by the inactivation of peptide YY (25). The other is the inhibitory effect
on fat absorption from the gut. Notably, another DPP-IV inhibitor,
vildagliptin, is reported to reduce the prandial triglyceride
response to fat-rich meal intake by 85% (26). Furthermore, sitagliptin in
combination with metformin reduce cholesterol and triglyceride
levels (27). In the present
study, since food consumption was unaltered between the two mouse
groups, the weight loss in the sitagliptin group was likely due to
the inhibition of fat absorption from the gut. Trials of weight
loss improve liver function and liver histology in NAFLD (28). The improvement of liver histology
by sitagliptin may originate from both weight-independent and
weight-dependent effects (29,30).
Treatment with sitagliptin activated PPAR-α and
attenuated SREBP1c and FAS. Therefore, the present study supported
that DPP-IV affected lipid accumulation by the inactivation of
PPAR-α, which is involved in β-oxidation of fatty acids, and the
activation of SREBP1c and FAS, which are involved in hepatic
lipogenesis (31). In the liver of
high-fat-fed mice lacking DPP-IV, the expression of PPAR-α is
upregulated, whereas the expression of SREBP1c is downregulated
(10). SREBP1c stimulates several
lipogenic enzymes, including FAS (32). In the present study, sitagliptin
inhibited FAS more markedly compared with SREBP1c. This may be
explained by the dose-dependency of sitagliptin (2 mg/kg/day).
Sitagliptin inhibits plasma DPP-IV activity in a dose-dependent
manner, from 0.1 to 3 mg/kg, in mice (33). Administration of a higher dose of
sitagliptin may reduce the expression of SREBP1c. MTP transports
triglycerides to very low-density lipoprotein. Enhanced expression
of this gene (34) promotes the
release of excess lipid from NAFLD livers. However, in the present
study, sitagliptin revealed no affect on MTP gene expression. From
these results, decreased steatosis by sitagliptin was caused by the
attenuation of lipogenesis and the stimulation of lipolysis.
The present study demonstrated that sitagliptin
reduced hepatic macrophages and Kupffer cells, which were measured
by the F4/80 positive cell index. Furthermore, the expression
levels of TNF-α and MCP-1 were reduced in the sitagliptin group.
Several chemokines are known to be target peptides of DPP-IV
(11). MCP-1 [chemokine (C-C
motif) ligand 2] is produced by Kupffer cells and HSCs, and
promotes hepatic inflammation by recruiting and activating
macrophages (35–37). The plasma concentration of MCP-1 is
reported to be reduced by treatment with DPP-IV inhibitors in
patients with diabetes (38).
DPP-IV inhibitors may reduce MCP-1 mediated C-C chemokine receptor
type 2 macrophages. From these results, sitagliptin induced a
marked reduction in intrahepatic inflammation.
In addition to the significant reduction of α-SMA
positive cells, activated myofibroblasts/HSCs, reduced hepatic
fibrosis area and decreased gene expression of procollagen I,
TGF-β1 and TIMP-1 were observed in the sitagliptin group compared
with the control group. Similarly, sitagliptin reduced the
activation of HSC in MCD fed mice (18). The mechanisms by which sitagliptin
attenuated fibrosis are most likely mediated through inhibition of
HSC activation. HSCs are activated by TGF-β1 or oxidative stress.
The present study immunochemically analyzed 8-OHdG and 4-HNE to
determine the effects of sitagliptin on oxidative stress. Oxidative
stress is pivotal as a factor in liver disease, which progresses
from steatosis to steatohepatitis. Fatty acid oxidation represents
an important source of reactive oxygen species, which induce lipid
peroxidation and initiate DNA damage, which are assessed as 4-HNE
and 8-OHdG production, respectively. It was demon-strated that
sitagliptin ameliorated 8-OHdG and 4-HNE immunostaining in the
liver tissues and attenuated reactive oxygen species production.
The present study revealed that sitagliptin suppressed the
activation of HSC via the attenuation of oxidative stress and the
inhibition of pro-fibrogenic and pro-inflammatory cytokines
(TGF-β1, TNF-α and MCP-1). Additionally, sitagliptin was reported
to directly suppress the proliferation of HSCs in rats (39). Therefore, sitagliptin may directly
and indirectly suppress the activation of HSCs.
In conclusion, sitagliptin attenuated the
progression of hepatic fibrosis by improving fatty deposition and
inhibiting inflammation. This treatment also decreased oxidative
stress, and pro-inflammatory and fibrogenic cyto-kines.
Abbreviations:
|
NASH
|
non-alcoholic steatohepatitis
|
|
DPP
|
dipeptidyl peptidase
|
|
NAFLD
|
on-alcoholic fatty liver disease
|
|
FLS
|
fatty liver shionogi
|
|
MCD
|
methionine/choline-deficient
|
|
AST
|
aminotransferase
|
|
ALT
|
alanine aminotransferase
|
|
HSC
|
hepatic stellate cell
|
|
SMA
|
α-smooth muscle actin
|
|
8-OHdG
|
8-hydroxy-2-deoxyguanosine
|
|
4-HNE
|
4-hydroxynonenal
|
|
TUNEL
|
terminal deoxynucleotidyl transferase
dUTP nick end labeling
|
|
PCR
|
polymerase chain reaction
|
|
TGF-β1
|
transforming growth factor-β1
|
|
CTGF
|
connective tissue growth factor
|
|
TNF-α
|
tumor necrosis factor-α
|
|
MCP-1
|
monocyte chemoattractant protein-1
|
|
TIMP-1
|
tissue inhibitor of
metalloproteinases-1
|
|
PPAR-α
|
peroxisome proliferator activated
receptor
|
|
SREBP1c
|
sterol regulatory element-binding
protein 1c
|
|
FAS
|
fatty acid synthase
|
|
MTP
|
microsomal triglyceride transfer
protein
|
References
|
1
|
Ding X, Saxena NK, Lin S, Gupta N and
Anania FA: Exendin-4, a glucagon-like protein-1 (GLP-1) receptor
agonist, reverses hepatic steatosis in ob/ob mice. Hepatology.
43:173–181. 2006. View Article : Google Scholar
|
|
2
|
Mentzel S, Dijkman HB, Van Son JP, Koene
RA and Assmann KJ: Organ distribution of aminopeptidase A and
dipeptidyl peptidase IV in normal mice. J Histochem Cytochem.
44:445–461. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Heike M, Möbius U, Knuth A, Meuer S and
Meyer zum Büschenfelde KH: Tissue distribution of the T cell
activation antigen Ta1. Serological, immunohistochemical and
biochemical investigations. Clin Exp Immunol. 74:431–434.
1988.PubMed/NCBI
|
|
4
|
Gorrell MD, Gysbers V and McCaughan GW:
CD26: A multi-functional integral membrane and secreted protein of
activated lymphocytes. Scand J Immunol. 54:249–264. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dinjens WN, ten Kate J, Wijnen JT, van der
Linden EP, Beek CJ, Lenders MH, Khan PM and Bosman FT: Distribution
of adenosine deaminase-complexing protein in murine tissues. J Biol
Chem. 264:19215–19220. 1989.PubMed/NCBI
|
|
6
|
Brubaker PL and Drucker DJ:
Structure-function of the glucagon receptor family of G
protein-coupled receptors: The glucagon, GIP, GLP-1 and GLP-2
receptors. Receptors Channels. 8:179–188. 2002. View Article : Google Scholar
|
|
7
|
Reinehr T, Roth CL, Enriori PJ and Masur
K: Changes of dipeptidyl peptidase IV (DPP-IV) in obese children
with weight loss: Relationships to peptide YY, pancreatic peptide
and insulin sensitivity. J Pediatr Endocrinol Metab. 23:101–108.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yamabe T, Takakura K, Sugie K, Kitaoka Y,
Takeda S, Okubo Y, Teshigawara K, Yodoi J and Hori T: Induction of
the 2B9 antigen/dipeptidyl peptidase IV/CD26 on human natural
killer cells by IL-2, IL-12 or IL-15. Immunology. 91:151–158. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ohnuma K, Takahashi N, Yamochi T, Hosono
O, Dang NH and Morimoto C: Role of CD26/dipeptidyl peptidase IV in
human T cell activation and function. Front Biosci. 13:2299–2310.
2008. View Article : Google Scholar
|
|
10
|
Conarello SL, Li Z, Ronan J, Roy RS, Zhu
L, Jiang G, Liu F, Woods J, Zycband E, Moller DE, et al: Mice
lacking dipeptidyl peptidase IV are protected against obesity and
insulin resistance. Proc Natl Acad Sci USA. 100:6825–6830. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Itou M, Kawaguchi T, Taniguchi E and Sata
M: Dipeptidyl peptidase-4: A key player in chronic liver disease.
World J Gastroenterol. 19:2298–2306. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Miyazaki M, Kato M, Tanaka K, Tanaka M,
Kohjima M, Nakamura K, Enjoji M, Nakamuta M, Kotoh K and Takayanagi
R: Increased hepatic expression of dipeptidyl peptidase-4 in
non-alcoholic fatty liver disease and its association with insulin
resistance and glucose metabolism. Mol Med Rep. 5:729–733.
2012.
|
|
13
|
Balaban YH, Korkusuz P, Simsek H, Gokcan
H, Gedikoglu G, Pinar A, Hascelik G, Asan E, Hamaloglu E and Tatar
G: Dipeptidyl peptidase IV (DDP IV) in NASH patients. Ann Hepatol.
6:242–250. 2007.PubMed/NCBI
|
|
14
|
Ben-Shlomo S, Zvibel I, Shnell M, Shlomai
A, Chepurko E, Halpern Z, Barzilai N, Oren R and Fishman S:
Glucagon-like peptide-1 reduces hepatic lipogenesis via activation
of AMP-activated protein kinase. J Hepatol. 54:1214–1223. 2011.
View Article : Google Scholar
|
|
15
|
Soga M, Kishimoto Y, Kawaguchi J, Nakai Y,
Kawamura Y, Inagaki S, Katoh K, Oohara T, Makino S and Oshima I:
The FLS mouse: A new inbred strain with spontaneous fatty liver.
Lab Anim Sci. 49:269–275. 1999.PubMed/NCBI
|
|
16
|
Soga M, Hashimoto S, Kishimoto Y, Hirasawa
T, Makino S and Inagaki S: Insulin resistance, steatohepatitis and
hepatocellular carcinoma in a new congenic strain of Fatty Liver
Shionogi (FLS) mice with the Lep (ob) gene. Exp Anim. 59:407–419.
2010. View Article : Google Scholar
|
|
17
|
Sugihara T, Koda M, Kishina M, Kato J,
Tokunaga S, Matono T, Ueki M and Murawaki Y: Fatty liver Shionogi
ob/ob mouse: A new candidate for a non-alcoholic steatohepatitis
model. Hepatol Res. 43:547–556. 2013. View Article : Google Scholar
|
|
18
|
Jung YA, Choi YK, Jung GS, Seo HY, Kim HS,
Jang BK, Kim JG, Lee IK, Kim MK and Park KG: Sitagliptin attenuates
methionine/choline-deficient diet-induced steatohepatitis. Diabetes
Res Clin Pract. 105:47–57. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
McKnight AJ, Macfarlane AJ, Dri P, Turley
L, Willis AC and Gordon S: Molecular cloning of F4/80, a murine
macrophage-restricted cell surface glycoprotein with homology to
the G-protein-linked transmembrane 7 hormone receptor family. J
Biol Chem. 271:486–489. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data usingreal-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
21
|
Tilg H and Moschen AR: Evolution of
inflammation in nonalcoholic fatty liver disease: The multiple
parallel hits hypothesis. Hepatology. 52:1836–1846. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tessari P, Coracina A, Cosma A and Tiengo
A: Hepatic lipid metabolism and non-alcoholic fatty liver disease.
Nutr Metab Cardiovasc Dis. 19:291–302. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Angulo P: Nonalcoholic fatty liver
disease. N Engl J Med. 346:1221–1231. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Youssef W and McCullough AJ: Diabetes
mellitus, obesity and hepatic steatosis. Semin Gastrointest Dis.
13:17–30. 2002.PubMed/NCBI
|
|
25
|
Ballantyne GH: Peptide YY(1–36) and
peptide YY(3–36): Part I. Distribution, release and actions. Obes
Surg. 16:651–658. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Matikainen N, Mänttäri S, Schweizer A,
Ulvestad A, Mills D, Dunning BE, Foley JE and Taskinen MR:
Vildagliptin therapy reduces postprandial intestinal
triglyceride-rich lipoprotein particles in patients with type 2
diabetes. Diabetologia. 49:2049–2057. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Scott R, Loeys T, Davies MJ and Engel SS;
Sitagliptin Study 801 Group: Efficacy and safety of sitagliptin
when added to ongoing metformin therapy in patients with type 2
diabetes. Diabetes Obes Metab. 10:959–969. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Schreuder TC, Verwer BJ, van Nieuwkerk CM
and Mulder CJ: Nonalcoholic fatty liver disease: An overview of
current insights in pathogenesis, diagnosis and treatment. World J
Gastroenterol. 14:2474–2486. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shirakawa J, Fujii H, Ohnuma K, Sato K,
Ito Y, Kaji M, Sakamoto E, Koganei M, Sasaki H, Nagashima Y, et al:
Diet-induced adipose tissue inflammation and liver steatosis are
prevented by DPP-4 inhibition in diabetic mice. Diabetes.
60:1246–1257. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Foley JE and Jordan J: Weight neutrality
with the DPP-4 inhibitor, vildagliptin: Mechanistic basis and
clinical experience. Vasc Health Risk Manag. 6:541–548. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Foufelle F and Ferré P: New perspectives
in the regulation of hepatic glycolytic and lipogenic genes by
insulin and glucose: A role for the transcription factor sterol
regulatory element binding protein-1c. Biochem J. 366:377–391.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bugianesi E, MuCullough AJ and Marchesini
G: Insulin resistance: A metabolic pathway to chronic liver
disease. Hepatology. 42:987–1000. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim D, Wang L, Beconi M, Eiermann GJ,
Fisher MH, He H, Hickey GJ, Kowalchick JE, Leiting B, Lyons K, et
al: (2R)-4-oxo-4-(3-(trifluoromethyl)-5,6-dihydro(1,2,4)
triazolo(4,3-a) pyrazin-7(8H)-yl)-1-(2,4,5-trifluorophenyl)
butan-2-amine: A potent, orally active dipeptidyl peptidase IV
inhibitor for the treatment of type 2 diabetes. J Med Chem.
48:141–151. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wetterau JR, Lin MC and Jamil H:
Microsomal triglyceride transfer protein. Biochim Biophys Acta.
1345:136–150. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zamara E, Galastri S, Aleffi S, Petrai I,
Aragno M, Mastrocola R, Novo E, Bertolani C, Milani S, Vizzutti F,
et al: Prevention of severe toxic liver injury and oxidative stress
in MCP-1-deficient mice. J Hepatol. 46:230–238. 2007. View Article : Google Scholar
|
|
36
|
Seki E, de Minicis S, Inokuchi S, Taura K,
Miyai K, van Rooijen N, Schwabe RF and Brenner DA: CCR2 promotes
hepatic fibrosis in mice. Hepatology. 50:185–197. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Galastri S, Zamara E, Milani S, Novo E,
Provenzano A, Delogu W, Vizzutti F, Sutti S, Locatelli I, Navari N,
et al: Lack of CC chemokine ligand 2 differentially affects
inflammation and fibrosis according to the genetic background in a
murine model of steatohepatitis. Clin Sci (Lond). 123:459–471.
2012. View Article : Google Scholar
|
|
38
|
Fadini GP, Boscaro E, Albiero M, Menegazzo
L, Frison V, de Kreutzenberg S, Agostini C, Tiengo A and Avogaro A:
The oral dipeptidyl peptidase-4 inhibitor sitagliptin increases
circulating endothelial progenitor cells in patients with type 2
diabetes: Possible role of stromal-derived factor-1alpha. Diabetes
Care. 33:1607–1609. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kaji K, Yoshiji H, Ikenaka Y, Noguchi R,
Aihara Y, Douhara A, Moriya K, Kawaratani H, Shirai Y, Yoshii J, et
al: Dipeptidyl peptidase-4 inhibitor attenuates hepatic fibrosis
via suppression of activated hepatic stellate cell in rats. J
Gastroenterol. 49:481–491. 2013. View Article : Google Scholar : PubMed/NCBI
|