Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the
fourth leading cause of cancer-associated mortality, with a
five-year survival rate of 5%. Surgery remains the most effective
treatment, however only 20% of patients are suitable for radical
resection (1).
Advances in chemotherapy, such as FOLFIRINOX and
gemcitabine plus nab-paclitaxel regimens, have resulted in an
improvement in outcomes, and gemcitabine-based chemotherapy remains
the gold-standard treatment, particularly in metastatic disease
(2–4).
Despite limited impact on patient care, recent PDAC
genomic characterization via the molecular dissection of somatic
alterations has generated informative data on different types of
cancer. However, advances in the development of novel PDAC
therapeutic and early-detection strategies remain to be identified.
The current study aimed to implement whole transcriptome massively
parallel sequencing (RNASeq) and copy number analysis to
investigate the molecular biology of PDAC, to aid in improving
diagnosis and indicating potential targets for personalized
diagnostic and therapeutic intervention. RNASeq is a powerful tool
for the identification of cancer mutations underlying pancreatic
carcinogenesis, and is an efficient approach for the detection of
somatic events such as nucleotide substitution mutations and gene
translocations with high resolution, via sequencing of the
expressed gene (cell transcriptomes). In addition, this technology
has the advantage of not being limited to known genes and
additionally may detect novel transcripts and alternative splice
forms (5).
The present study combined RNASeq and copy number
analysis by the single-nucleotide polymorphism (SNP) array to
obtain a comprehensive overview of genetic alterations in
pancreatic cancer.
Patients and methods
The current study was conducted according to the
principles of the Declaration of Helsinki, and written informed
consent was obtained from all participants. The study was
previously approved by the Independent Ethics Committee of
Sant'Orsola-Malpighi Hospital (Bologna, Italy).
Sample collection and patient
characteristics
A total of 24 PDAC samples were collected from
ultrasound-guided biopsies or surgical specimens for DNA and RNA
extraction, however 16 specimens were analyzed due to the exclusion
of 3 cases for non-PDAC histology, 2 cases for insufficient nucleic
acid extraction and 3 cases for estimated low cellularity. Patient
characteristics are presented in Table
I. The tissue samples were collected in cryogenic tubes (Ambion
Life Technologies, Carlsbad, CA, USA) and stored at −20°C in
RNAlater solution (Ambion Life Technologies). Nucleic acid
extraction was performed with the AllPrep RNA/DNA kit (Qiagen,
Inc., Valencia, CA, USA) for tumor biopsies and the QIAAmp DNA Mini
kit (Qiagen, Inc.) for peripheral blood DNA.
 | Table ICharacteristics of the patient cohort
with a median age of 65.5 years (range, 34–87). |
Table I
Characteristics of the patient cohort
with a median age of 65.5 years (range, 34–87).
| Characteristic | % of study
population |
|---|
| Gender | |
| Male | 23.5 |
| Female | 76.5 |
| Site of specimen | |
| Pancreatic
tumor | 94 |
| Hepatic
metastasis | 6 |
| Stage | |
| I+II | 41 |
| III-LA | 29.5 |
| IV | 29.5 |
Sample cellularity
PDAC is characterized by a small quantity of
adenocarcinoma cells (6).
Evaluation of tumor cells in the sample was based on the presence
and relative enrichment of the Kirsten rat sarcoma viral oncogene
homolog (KRAS) mutation. The percentage of tumor cells in the
clinical specimen was estimated using KRAS mutation Sanger
sequencing. Samples with 10% more tumor alleles than normal alleles
were included in the present study.
Copy number alteration analysis
Tumor DNA was labeled and hybridized to GeneChip SNP
6.0 arrays (Affymetrix, Inc., Santa Clara, CA, USA) following the
manufacturer's instructions. Copy number analysis was conducted
using Partek Genomic Suite software (version 6.5; Partek, Inc., St.
Louis, MO, USA), using the segmentation algorithm and setting the
parameters to identify large and cryptic regions of copy number
alterations (P-value =0.001; number of markers =10; signal to noise
ratio =0.3). The copy number was assessed by comparing the
intensity distribution to a reference set consisting of
approximately 270 normal samples from individuals of different
ethnicities derived from the HapMap database (http://hapmap.ncbi.nlm.nih.gov/).
Whole transcriptome massively parallel
sequencing
Following RNA extraction using the AllPrep RNA/DNA
kit (Qiagen, Inc.,), whole transcriptome sequencing was performed
on the Illumina HiScanSQ platform, (Illumina, Inc., San Diego, CA,
USA) using Illumina paired-end massively parallel sequencing in
accordance with the manufacturer's instructions. Poly(A)-RNA was
purified from 250–500 ng total RNA using poly-T oligo-attached
magnetic beads (Illumina, Inc.), and libraries of cDNA fragments
were sequenced at 2×80 base pairs (bp) read length in paired end
mode. An average of 98.2 million reads/sample were produced.
For exome sequencing, genomic DNA from peripheral
blood was fragmented, tagged, indexed and amplified with the
Nextera Exome Enrichment kit (Illumina, Inc.) to an average library
size of 350 bp. DNA libraries were hybridized to biotin-labeled
95-mer probes (Illumina, Inc.) designed to enrich 62 Mb of genomic
DNA covering more than 200,000 exons including exon-flanking
regions. Sequencing was performed at 100 bp in the paired end,
producing on average 2.6 Gb per sample.
For bioinformatics analysis, the short reads were
processed, quality-filtered and mapped on the human reference
genome to identify all detectable variations in the sample,
including single-nucleotide variants (SNVs), insertions or
deletions (In/Dels) and large chromosomal rearrangements.
Open-source software [BowTie (http://bowtie-bio.sourceforge.net/index.shtml), TopHat
(https://ccb.jhu.edu/software/tophat/index.shtml),
SAMtools (http://samtools.sourceforge.net/) and SNVMix
(http://compbio.bccrc.ca/software/snvmix/)] was used
for mapping. Novel variants were identified by comparison with
public databases on human variability [1000 Genomes (http://www.1000genomes.org/), dbSNP (http://www.ncbi.nlm.nih.gov/SNP/) and Cosmic
(http://cancer.sanger.ac.uk/cosmic)]
and the potential effect of the non-synonymous SNVs was evaluated
at a protein level with computational tools including SNPs&GO
(http://snps-and-go.biocomp.unibo.it/snps-and-go/),
SIFT (http://sift.jcvi.org/) and PolyPhen
(http://genetics.bwh.harvard.edu/pph2/). deFuse
(http://compbio.bccrc.ca/software/defuse/), ChimeraScan
(https://code.google.com/p/chimerascan/) and FusionMap
(http://www.array-server.com/wiki/index.php?title=FusionMap)
were used to identify chromosomal rearrangements resulting in gene
fusion.
Sanger sequencing
SNVs were confirmed by Sanger sequencing on tumor
samples and peripheral blood DNA (when available) on an ABI 3730
Genetic Analyzer (Applied Biosystems Life Technologies, Monza,
Italy). Primer pairs, designed with Primer Express software,
version 3.0 (Applied Biosystems Life Technologies), were specific
to amplify exons and the flanking intronic regions. PCR products
were purified with the QIAquick PCR purification kit (Qiagen, Inc.)
and sequenced on both strands using the BigDye Terminator v1.1
Cycle Sequencing kit (Applied Biosystems Life Technologies).
Results
Whole transcriptome sequencing of the 16 samples
derived from surgical specimens or ultrasound-guided biopsies was
conducted. Of these, 13 specimens additionally underwent copy
number analysis using Affymetrix technology to obtain a
comprehensive overview of the genetic alterations in pancreatic
cancer.
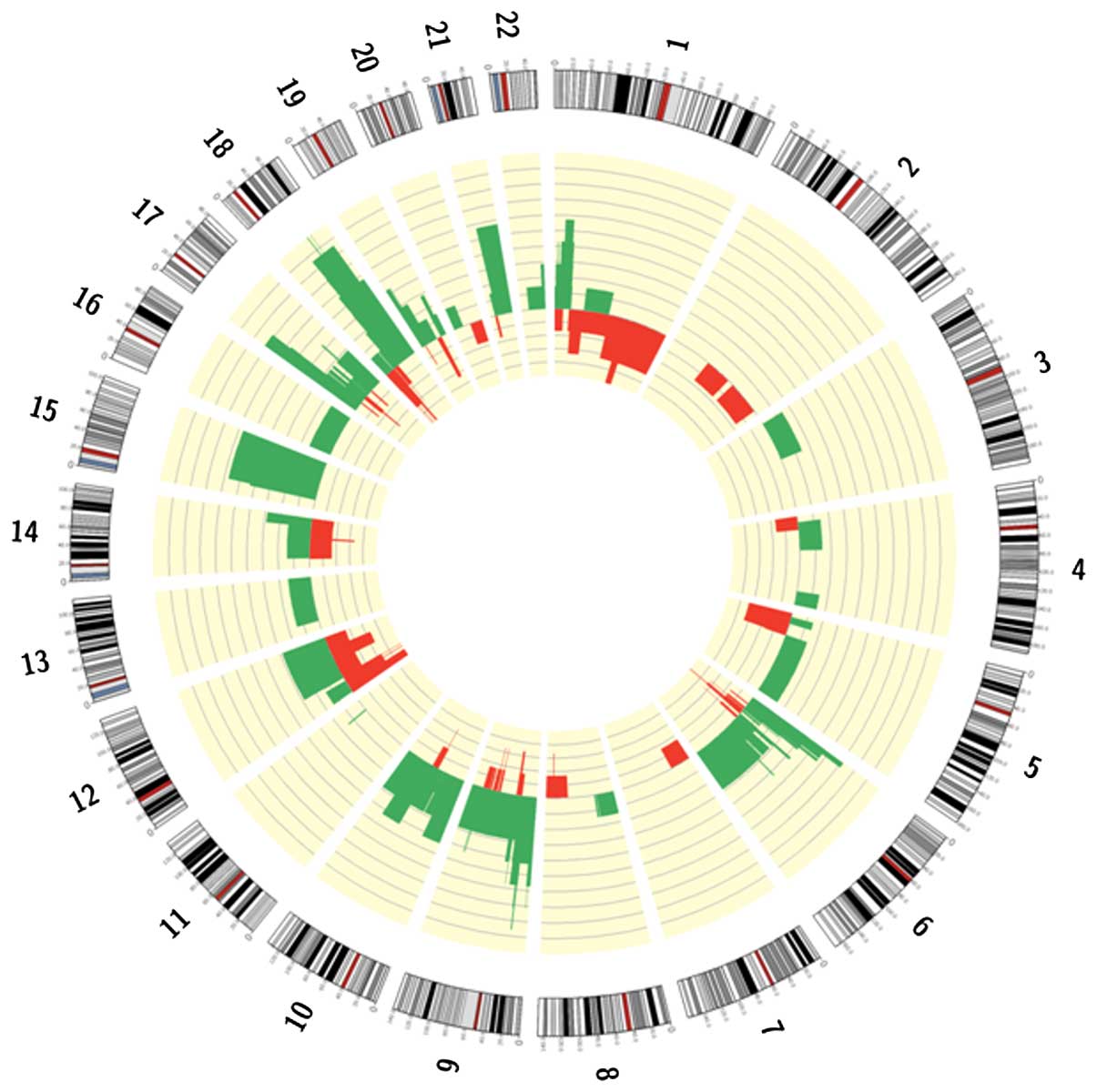
SNP array identified focal or macroscopic
amplifications and deletions in 85% of patients (Fig. 1). Gains frequently involved
chromosome arms 12p, 18q, 19q and 6p, which contain the oncogenes
KRAS, GATA binding protein 6 (GATA6), protein kinase B (AKT2) and
cyclin D3 (CCND3). Focal high copy number amplifications were
identified in the 8q24 region involving the MYC gene, and in 17q12
surrounding human epidermal growth factor receptor 2 (ERBB2) in one
and two patients, respectively. Deletions were observed in greater
than 70% of patients on chromosome arms 1p, 9p, 6p, 18q, 10q, 15q,
17p, 21q and 19p. Deleted genes were commonly those previously
associated with PDAC, including tumor protein 53 (TP53),
cyclin-dependent kinase inhibitor 2A (CDKN2A/B) and mothers against
deca-pentaplegic homolog 4 (SMAD4). In addition, runt-related
transcription factor 2, AT-rich interactive domain-containing
protein 1A (ARID1A), phosphatase and tensin homolog (PTEN)and
serine/threonine kinase 11 were identified to be deleted. An
additional recurrent deletion involved chromosome 15q, however a
specific target oncosuppressor has not been identified in this
region at present.
RNASeq identified an average of 264 coding
non-synonymous novel SNVs (ranging from 146–374) and 16 novel
In/Dels (ranging from 6–24) for each sample, of which a mean of
11.2% (from 4.8–17.6%) were disease-associated and somatic events
and 34.7% (from 13.3–52.2%) were frame-shift somatic In/Dels (for
the patient with matched-normal DNA available). Furthermore, gene
fusions were detected in 10 patients with a total of 23 different
intra- or inter-chromosomal rearrangements, however the recurrent
fusion transcript was not identified. The majority of
rearrangements were inter-chromosomal (66%) and did not alter the
reading-frame (51%).
KRAS was reported to have the greatest prevalence of
somatic mutations (93.7% of cases) and the mutations affected the
known hotspot at codon 12 (G12D in 8 patients, G12V and G12R in 5
and 2 patients respectively) (7).
In one case which was negative for KRAS, a G13D neuroblastoma RAS
viral oncogene homolog (NRAS) mutation was identified. The
prevalence of the RAS mutation in the current study is biased due
to the experimental design, as samples were only included in the
study if a mutation in a RAS gene was present. This strategy was
designed to take into account the common occurrence of stromal
desmoplasia in the majority of cases of PDAC that may affect the
percentage of tumor cells present in biopsies or surgical specimens
undergoing analysis. The percentage of tumor cells in the sample
were estimated by quantifying the RAS mutation in the DNA, taking
into account copy number gains or the loss of chromosome 12. This
analysis enabled the inclusion of 16 patients (out of the 20
initially assayed) that exhibited an average of 41.9±7.48% tumor
cells in the sample.
The most recurrent genes observed to be altered by
point mutations, small In/Dels and heterozygous or homozygous copy
number loss are presented in Fig.
2, with TP53, CDKN2A and SMAD4 observed to be commonly
inactivated genes. TP53 was inactivated in 56% of tumor samples due
to somatic point mutations (5/16) or allelic loss (5/16), while
CDKN2A function was disrupted in 8 of the 16 samples by missense or
nonsense mutations and hetero/homozygous genomic loss. TP53 and
CDKN2A alterations were frequently coupled, with 6 of the 11
patients exhibiting mutations in these two genes. Transforming
growth factor-β (TGF-β) signaling was impaired in 50% of patients,
frequently due to hetero- or homozygous focal loss of the SMAD4
locus. In addition, SMAD3 was frequently inactivated by point
mutation, gene fusion or allelic loss. Somatic mutations in the
additional genes identified had reduced ocurrence, compared with
the frequency of KRAS, SMAD4, TP53 and CDKN2A. However, an
exception was ARID1A, the inactivation of which was observed in 6
of the 16 patients due to heterozygous focal genomic deletions or
frameshift small In/Dels. In addition, the known oncosuppressor
gene, PTEN, was inactivated due to nonsense or missense mutations,
or chromosome arm deletion. Notably, PTEN alterations were more
frequent in patients with advanced disease (4/8) compared with
early stage subjects (0/7).
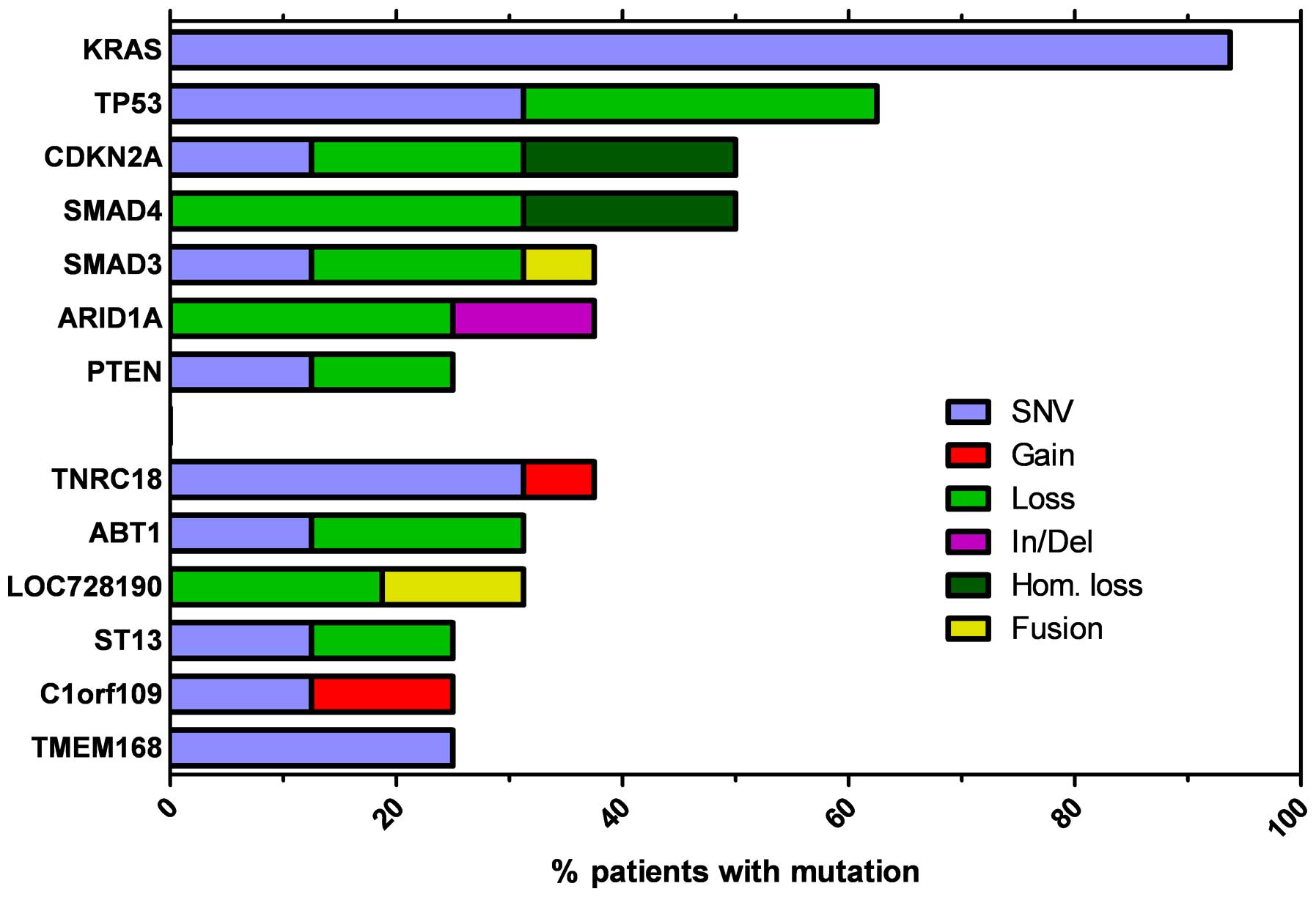 | Figure 2Most recurrent genes altered by point
mutation, small In/Del and heterozygous or homozygous copy number
loss. KRAS, Kirsten rat sarcoma viral oncogene homolog; TP53, tumor
protein 53; CDKN2A, cyclin-dependent kinase inhibitor 2A; SMAD4,
mothers against decapentaplegic homolog 4; ARID1A, AT-rich
interactive domain-containing protein 1A; PTEN, phosphatase and
tensin homolog; TNRC18, trinucleotide repeat containing 18; ABT1,
activator of basal transcription 1; LOC728190, locus 728190; ST13,
suppression of tumorigenicity 13; C1orf109, chromosome 1 open
reading frame 109; TMEM168, transmembrane protein 168; SNV,
single-nucleotide variant; In/Del, insertion or deletion; Hom.
loss, loss of homozygosity. |
The DNA damage response pathway is significantly
impaired in pancreatic tumorigenesis (8). Pathway analysis of the genes carrying
somatic alterations in a minimum of two patients exhibted a
significant enrichment in DNA repair mechanisms (P=0.007, fold
enrichment = 9.6). Of note, mismatch repair, base-excision and
nucleotide excision repair pathways were altered in different
patients. In Fig. 2 the most
recurrent genes altered by point mutations, small In/Dels and
heterozygous or homozygous copy number loss are presented.
Discussion
Pancreatic cancer presents genetic heterogeneity
with a high number of mutations occurring in single cases. Four
genes with a high prevalence of mutations have been identified:
KRAS mutation, CDKN2A inactivation (considered to be early events
in the PDAC progression model), TP53 and SMAD4 inactivation
(considered as later events) (9,10).
The majority of single gene mutations in pancreatic
cancer are grouped into common cellular pathways. Jones et
al (11) identified 69 mutated
gene sets in the majority of the 24 samples analyzed, of which 31
were grouped into 12 core signaling pathways. These pathways
included KRAS, TGF-β, DNA damage control, apoptosis and regulation
of G1/S cell cycle transition, involving KRAS, CDKN2A,
TP53 and SMAD4. In addition, pathways including Hedgehog signaling,
the homophilic cell adhesion pathway, integrin signaling, TGF-β
signaling, Wnt/Notch signaling, and regulation of the invasion
pathway were identified (11).
The current study confirmed the high prevalence of
KRAS, CDKN2A, TP53 and SMAD4 mutations. In particular, 93.7% of
tumor samples exhibited somatic mutations activating KRAS and gene
amplifications. Although the high KRAS mutation prevalence is an
experimental design bias, as samples were included in the study
only if a mutation in any RAS gene was observed, a previous study
indicated a similar mutation frequency (12).
Notably, a G13D NRAS mutation was identified in the
one case negative for KRAS. Ras proteins are GTPases with a high
frequency of mutations in human tumors, however KRAS isoform
mutations are prominent in pancreatic cancer compared with
mutations of NRAS isoforms, which are more common in malignant
melanomas (13).
KRAS is a key oncogene during the onset of
pancreatic cancer however an effective KRAS inhibitor remains to be
identified (14). SNP array
technologies in the present study indicated amplifications at the
chromosome arms harboring additional oncogenic genes such as GATA6,
MYC and AKT2. In particular, AKT2 is involved in the
phosphoinositide 3-kinase (PI3K) pathway, one of the feedback
mechanisms through which KRAS maintains its levels of activity
(15). Williams et al
(16) observed that the combined
use of mitogen-activated protein kinase (MAPK) and AKT inhibitors
increased tumor sensitivity to radiation in a xenograft model of
pancreatic cancer, while Diersch et al (15) demonstrated how PI3K inhibition
reduced proliferation and tumor growth in a KPC mouse model of
PDAC.
Therefore inhibition of PI3K or of additional
pathways activated downstream of KRAS, such as MAPK signaling, may
be an alternative way to target KRAS in PDAC (7).
The current study observed SMAD4 inactivation due to
hetero- or homozygous focal loss of the SMAD4 locus (18q21.2)
involved in the TGF-β pathway. SMAD family proteins involved in the
TGF-β pathway were organized into two gene clusters, one at 18q21
chromosome (SMAD2, SMAD4 and SMAD7) and one at 15q21–22 (SMAD3 and
SMAD6) (17).
TGF-β receptor 1 activates SMAD2 and SMAD3, which
bind to the common partner SMAD4, while SMAD6 and SMAD7 have an
inhibitory role and block the phosphorylation of SMAD2 or SMAD3
(18). The SMAD complex regulates
the transcription of several TGF-β-dependent genes following
nuclear translocation that may have a context-dependent, tumor
suppressive or progressive role (18). However, in the current study SMAD4
inactivation was observed at a reduced frequency than reported in a
previous study (19).
Hezel et al (20) reported a 90% of loss of
heterozygosity at the SMAD4 locus in PDAC, 50% of which exhibited
an additional inactivation of the remaining allele. In the current
study, 50% of SMAD4 inactivation was observed to be due to hetero-
and homozygous focal loss of the SMAD4 locus (18q21.2) in 62.5 and
37.5% of SMAD4 inactivation samples, respectively. Notably,
patients with homozygous loss of SMAD4 (37.5% of SMAD4 mutation
cases) exhibited a poorer prognosis and a more aggressive disease.
Leung et al (21)
demonstrated that SMAD4 expression suppressed PDAC metastasis in
their orthotopic xenograft model and Blackford et al
(22) reported that SMAD4 loss is
associated with a worsened PDAC prognosis. Therefore, SMAD4 is
suggested as a potential prognostic biomarker, which may aid in
development of therapeutic strategies (23). However, the current study
identified cases negative for SMAD4 mutations with SMAD3
inactivation due to point mutation, gene fusion or allelic loss,
with a possible role in pancreatic carcinogenesis in 37.5%.
The present study confirmed the important role in
pancreatic carcinogenesis of the oncosuppressor genes TP53 and
CDKN2A, however in addition PTEN inactivation was observed in 25%
of samples and more frequently in advanced patients (4/8) compared
with early stage subjects (0/7). PTEN, a negative regulator of the
PI3K pathway, may be involved in the gain of metastatic potential
of pancreatic tumor cells in the absence of SMAD4. Garcia-Carracedo
et al (24) investigated
the role of PI3K signaling dysregulation in intraductal papillary
mucinous neoplasm (IPMN), and loss of heterozygosity status at PTEN
was observed in 35.7% of the IPMN cases analyzed. This indicated
that PTEN down-regulation was associated with a poor prognosis in
patients with IPMN (24).
The current study demonstrated a high frequency of
alterations in genes involved in DNA damage repair and chromatin
remodeling. This highlights the relevance of the impaired DNA
damage response in pancreatic carcinogenesis, in agreement with
previous studies (8,25,26).
Dong et al (27) reported
that mismatch repair gene variants may affect susceptibility to
pancreatic cancer, observing that 28 SNPs were associated with
altered pancreatic cancer risk (P<0.05).
In addition, the present study demonstrated ARID1A
alterations in 6 of the 16 patients due to heterozygous focal
genomic deletions or small frameshift In/Dels. ARID1A is a gene
involved in DNA repair through the ATP-dependent induction of
chromatin migration and dissociation, and its loss is frequently
observed in ovarian clear cell adenocar-cinoma and endometrioid
adenocarcinoma (28). A previous
study indicated that ARID1A loss is associated with reduced
disease-free survival and chemoresistance in ovarian clear cell
adenocarcinoma, however ARID1A inactivation has been identified in
different tumor types including pancreatic cancer (29–31).
Additionally, the current study observed mutations
in genes with no known function, the most common being detected at
trinucleotide repeat containing 18, locus 728190, poliovirus
receptor related immunoglobulin domain, SH3-domain binding protein
2, transmembrane protein 168, DEAD box protein 60 and NHL repeat
containing 2. Further studies are required to investigate the
function and role of these gene mutations in pancreatic
carcinogenesis.
In conclusion, the present study confirmed the
tumoral heterogeneity of PDAC and identified known mutations in
genes involved in RAS signaling, the p53 pathway and TGF-β
signaling, including SMAD4 and SMAD3 mutations. In addition, an
emerging role for PTEN and ARID1A was identified and the importance
of impaired DNA damage repair in creating the genetic instability
responsible for cancer progression was emphasized.
Acknowledgments
The present study was supported by PRIN 2009 New
Therapeutic Strategies in Pancreatic Cancer and the Programma di
Ricerca Regione-Università, Regione Emilia Romagna, bando Giovani
Ricercatori ʻAlessandro Liberatiʼ 2013 to SV (no.
PRUA1GR-2013–00000038). The authors would like to thank the
Interdepartmental Center of Cancer Research for the technical
support.
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Conroy T, Desseigne F, Ychou M, Bouché O,
Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de
la Fouchardière C, et al: FOLFIRINOX versus gemcitabine for
metastatic pancreatic cancer. N Engl J Med. 364:1817–1825. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Von Hoff DD, Ervin T, Arena FP, Chiorean
EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, et
al: Increased survival in pancreatic cancer with nab-paclitaxel
plus gemcitabine. N Engl J Med. 369:1691–1703. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Di Marco M, Di Cicilia R, Macchini M,
Nobili E, Vecchiarelli S, Brandi G and Biasco G: Metastatic
pancreatic cancer: Is gemcitabine still the best standard
treatment? (Review) Oncol Rep. 23:1183–1192. 2010.
|
|
5
|
Meyerson M, Gabriel S and Getz G: Advances
in understanding cancer genomes through second-generation
sequencing. Nat Rev Genet. 11:685–696. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Feig C, Gopinathan A, Neesse A, Chan DS,
Cook N and Tuveson DA: The pancreas cancer microenvironment. Clin
Cancer Res. 18:4266–4276. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Collins MA and Pasca di Magliano M: Kras
as a key oncogene and therapeutic target in pancreatic cancer.
Front Physiol. 4:4072014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tan XG, Yang ZL, Yang LP and Miao XY:
Expression of DNA-repair proteins and their significance in
pancreatic cancer and non-cancerous pancreatic tissues of
Sprague-Dawley rats. World J Surg Oncol. 12:322014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hruban RH, Goggins M, Parsons J and Kern
SE: Progression model for pancreatic cancer. Clin Cancer Res.
6:2969–2972. 2000.PubMed/NCBI
|
|
10
|
Iacobuzio-Donahue CA, Velculescu VE,
Wolfgang CL and Hruban RH: Genetic basis of pancreas cancer
development and progression: insights from whole-exome and
whole-genome sequencing. Clin Cancer Res. 18:4257–4265. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jones S, Zhang X, Parsons DW, Lin JC,
Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, et
al: Core signaling pathways in human pancreatic cancers revealed by
global genomic analyses. Science. 321:1801–1806. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yachida S, White CM, Naito Y, Zhong Y,
Brosnan JA, Macgregor-Das AM, Morgan RA, Saunders T, Laheru DA,
Herman JM, et al: Clinical significance of the genetic landscape of
pancreatic cancer and implications for identification of potential
long-term survivors. Clin Cancer Res. 18:6339–6347. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fernández-Medarde A and Santos E: Ras in
cancer and developmental diseases. Genes Cancer. 2:344–358. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Takashima A and Faller DV: Targeting the
RAS oncogene. Expert Opin Ther Targets. 17:507–531. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Diersch S, Wenzel P, Szameitat M, Eser P,
Paul MC, Seidler B, Eser S, Messer M, Reichert M, Pagel P, et al:
Efemp1 and p27(Kip1) modulate responsiveness of pancreatic cancer
cells towards a dual PI3K/mTOR inhibitor in preclinical models.
Oncotarget. 4:277–288. 2013.PubMed/NCBI
|
|
16
|
Williams TM, Flecha AR, Keller P, Ram A,
Karnak D, Galbán S, Galbán CJ, Ross BD, Lawrence TS, Rehemtulla A
and Sebolt-Leopold J: Cotargeting MAPK and PI3K signaling with
concurrent radiotherapy as a strategy for the treatment of
pancreatic cancer. Mol Cancer Ther. 11:1193–1202. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Massagué J: TGFbeta in Cancer. Cell.
134:215–230. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Javle M, Li Y, Tan D, Dong X, Chang P, Kar
S and Li D: Biomarkers of TGF-β signaling pathway and prognosis of
pancreatic cancer. PLoS One. 9:e859422014. View Article : Google Scholar
|
|
19
|
Chow JY, Dong H, Quach KT, Van Nguyen PN,
Chen K and Carethers JM: TGF-beta mediates PTEN suppression and
cell motility through calcium-dependent PKC-alpha activation in
pancreatic cancer cells. Am J Physiol Gastrointest Liver Physiol.
294:G899–G905. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hezel AF, Kimmelman AC, Stanger BZ,
Bardeesy N and Depinho RA: Genetics and biology of pancreatic
ductal adeno-carcinoma. Genes Dev. 20:1218–1249. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Leung L, Radulovich N, Zhu CQ, Wang D, To
C, Ibrahimov E and Tsao MS: Loss of canonical Smad4 signaling
promotes KRAS driven malignant transformation of human pancreatic
duct epithelial cells and metastasis. PLoS One. 8:e843662013.
View Article : Google Scholar
|
|
22
|
Blackford A, Serrano OK, Wolfgang CL,
Parmigiani G, Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ,
Eshleman JR, et al: SMAD4 gene mutations are associated with poor
prognosis in pancreatic cancer. Clin Cancer Res. 15:4674–4679.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Oshima M, Okano K, Muraki S, Haba R, Maeba
T, Suzuki Y and Yachida S: Immunohistochemically detected
expression of 3 major genes (CDKN2A/p16, TP53, and SMAD4/DPC4)
strongly predicts survival in patients with resectable pancreatic
cancer. Ann Surg. 258:336–346. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Garcia-Carracedo D, Turk AT, Fine SA,
Akhavan N, Tweel BC, Parsons R, Chabot JA, Allendorf JD, Genkinger
JM, Remotti HE and Su GH: Loss of PTEN expression is associated
with poor prognosis in patients with intraductal papillary mucinous
neoplasms of the pancreas. Clin Cancer Res. 19:6830–6841. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lovejoy CA, Li W, Reisenweber S, Thongthip
S, Bruno J, de Lange T, De S, Petrini JH, Sung PA, Jasin M, et al:
ALT Starr Cancer Consortium: Loss of ATRX, genome instability, and
an altered DNA damage response are hallmarks of the alternative
lengthening of telomeres pathway. PLoS Genet. 8:e10027722012.
View Article : Google Scholar
|
|
26
|
Waddell N, Pajic M, Patch AM, Chang DK,
Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, et al:
Australian Pancreatic Cancer Genome Initiative: Whole genomes
redefine the mutational landscape of pancreatic cancer. Nature.
518:495–501. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dong X, Li Y, Hess KR, Abbruzzese JL and
Li D: DNA mismatch repair gene polymorphisms affect survival in
pancreatic cancer. Oncologist. 16:61–70. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hargreaves DC and Crabtree GR:
ATP-dependent chromatin remodeling: Genetics, genomics and
mechanisms. Cell Res. 21:396–420. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Katagiri A, Nakayama K, Rahman MT, Rahman
M, Katagiri H, Nakayama N, Ishikawa M, Ishibashi T, Iida K,
Kobayashi H, et al: Loss of ARID1A expression is related to shorter
progression-free survival and chemoresistance in ovarian clear cell
carcinoma. Mod Pathol. 25:282–288. 2012.
|
|
30
|
Biankin AV, Waddell N, Kassahn KS, Gingras
MC, Muthuswamy LB, Johns AL, Miller DK, Wilson PJ, Patch AM, Wu J,
et al: Australian Pancreatic Cancer Genome Initiative: Pancreatic
cancer genomes reveal aberrations in axon guidance pathway genes.
Nature. 491:399–405. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jones S, Li M, Parsons DW, Zhang X,
Wesseling J, Kristel P, Schmidt MK, Markowitz S, Yan H, Bigner D,
et al: Somatic mutations in the chromatin remodeling gene ARID1A
occur in several tumor types. Hum Mutat. 33:100–103. 2012.
View Article : Google Scholar
|