Introduction
Chronic lymphocytic leukemia (CLL) is the most
prevalent type of adult leukemia in western countries (1). Failure to eliminate residual leukemia
cells, which are resistant to drug treatment, and the eventual
reemergence of the leukemia cell population continue to be major
clinical challenges, and CLL remains an incurable disease (2). Although several anticancer drugs are
effective at eliminating CLL cells in vitro, leukemia cells
are much more resistant to drug treatment in vivo.
Increasing evidence suggests that the bone marrow stroma may
provide a tissue microenvironment, which promotes the survival of
CLL cells and facilitates drug resistance (3,4).
Therefore, the development of novel therapeutic strategies to
inhibit the protective effect of stromal cells is critical for the
effective elimination of malignant cells in vivo.
CLL cells are under intrinsic oxidative stress and
exhibit high spontaneous apoptosis with rapid glutathione (GSH)
depletion in vitro (5–7). GSH
is important in CLL cells, counteracting oxidative stress and
maintaining the redox balance (8).
By relieving oxidative stress, GSH also reduces the activity of
reactive oxygen species (ROS)-generating drugs (9). Our previous study revealed that bone
marrow stromal cells convert cystine to cysteine, allowing CLL
cells to synthesize GSH (8). This
metabolic interaction between CLL cells and bone marrow stromal
cells increases the expression levels of GSH in CLL cells, and
promotes cell survival. Interruption of this biochemical
interaction using the GSH-depletion agent, β-phenylethyl
isothiocyanate (PEITC), significantly sensitizes CLL cells to drug
treatment in the stromal environment (8). Therefore, PEITC is a potent candidate
for the development of combination treatment strategies to overcome
microenvironment-mediated drug resistance in CLL cells.
Histone deacetylase inhibitors (HDACIs) are emerging
as a potent novel class of anticancer agents (10). A previous study demonstrated that
HDACI triggers apoptosis via the intrinsic apoptotic signaling
pathway following early generation of ROS in acute myeloid leukemia
(AML) cell lines, and inhibition of ROS generation protects
leukemia cells from apoptosis (11). Our previous study suggested that
HDACI-induced ROS generation leads to the upregulation of
GSH-associated enzymatic genes in myeloid leukemia cells, and
confers resistance to HDACI toxicity (12). Therefore, the redox status of
malignant cells affects HDACI sensitivity, and modulating ROS
levels is important for the design of drug combination strategies
to overcome HDACI resistance.
The HDACI suberoylanilide hydroxamic acid (SAHA or
Vorinostat) is the first HDACI to be approved for use in the
treatment of cutaneous T-cell lymphoma (13). Preclinical studies have reported
that SAHA exerts promising antitumor activity in CLL cells
(14–16). However, initial monotherapy
clinical trials using various HDACIs in patients with CLL exhibited
limited efficacy (17,18), which indicates that the leukemia
microenvironment in vivo may affect drug sensitivity. The
mechanisms underlying the role of SAHA in CLL cells remains to be
elucidated, particularly in the context of
microenvironment-mediated redox changes in CLL cells. The aims of
the present study were to examine the role of ROS generation in
SAHA toxicity in CLL cells, to investigate the significance of bone
marrow stromal cell-mediated redox changes in protection against
SAHA-induced ROS stress and cell death in CLL cells, to evaluate
the effect of SAHA in combination with the PEITC redox-modulating
compound, and to determine its ability to eliminate
stromal-protected CLL cells.
Materials and methods
Reagents
SAHA, PEITC, N-acetylcysteine (NAC), metaphosphoric
acid, propidium iodide (PI), anti-β-actin, paraformaldehyde, Triton
X-100 and bovine serum albumin (BSA) were purchased from
Sigma-Aldrich (St. Louis, MO, USA). CM-H2DCF-DA, nonyl
acridine orange (NAO), Rhodamine-123 and mounting medium,
supplemented with 4′,6-diamidino-2-phenylindole (DAPI), were
purchased from Invitrogen Life Technologies (Carlsbad, CA, USA).
The Annexin V-fluorescein isothiocyanate (FITC), Z-VAD, a caspase-3
activity assay kit and recombinant active caspase-3 were purchased
from BD Biosciences (San Jose, CA, USA). Ficoll-lite Lympho H was
purchased from Atlanta Biologicals, Inc. (Flowery Branch, GA, USA).
(S)-4-carboxyphenylglycine (CPG) was acquired from Tocris
Bioscience (Ellisville, MO, USA). The GSH assay kit was purchased
from Cayman Chemical Company (Ann Arbor, MI, USA). Rabbit
anti-human γ-glutamyl cysteine synthetase (GCLC; cat. no.
sc-28965), rabbit anti-human nuclear factor-E2-related factor 2
(Nrf2; cat. no. sc-13032), and rabbit anti-human myeloid cell
leukemia 1 (Mcl1; cat. no. sc-819) were purchased from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA). Sealed modular incubator
chambers were purchased from Billups-Rothenberg, Inc. (San Diego,
CA, USA).
Cell lines and primary CLL cells
The HS5 human bone marrow stromal cell line
immortalized by E6/E7 (11), was
obtained from American Type Culture Collection (Manassas, VA, USA).
A total of 62 patients (male and female; aged 38–85 years)
diagnosed with typical B-CLL were recruited from the First
Affiliated Hospital of Nanchang University (Nanchang, China) in the
present study. The diagnosis was based on clinical criteria and
laboratory features, according to National Cancer Institute
Criteria (19). All patients
provided written informed consent, and the present study was
approved by the ethics committee of The First Affiliated Hospital
of Nanchang University. In all experiments, CLL cells were isolated
from peripheral blood samples, which were collected from the
patients, by density gradient centrifugation (20). Briefly, 5 ml blood was slowly added
to a Falcon tube containing 6 ml pre-warmed Fico/Lite LymphoH
buffer (Atlanta Biological, Lawrenceville, GA, USA) and then
centrifuged at 435 × g for 20 min at room temperature. The isolated
CLL cells was washed with pre-warmed PBS and incubated in RPMI-1640
medium (Cellgro; Mediatech, Inc., Hendon, VA, USA) supplemented
with 10% fetal bovine serum, penicillin (100 U/ml) and streptomycin
(100 µg/ml; all from Cellgro; Mediatech, Inc.) overnight
prior to experimentation.
Cell viability assays
The effect of bone marrow stromal cells on
drug-induced apoptosis in CLL cells was determined, as previously
described (8). Briefly, HS5 cells
(5×104 cells/ml) were seeded in 24-well plates and
allowed to adhere and grow overnight prior to the addition of CLL
cells. The CLL cells were isolated from the blood samples and
incubated overnight with RPMI-1640 medium supplemented with 10%
fetal bovine serum, penicillin (100 U/ml) and streptomycin (100
µg/ml), and then transferred (1×106) into 24-well
plates with or without the pre-seeded stromal cells at a density of
5×104 cells per well. Following co-culture for 1 day,
the CLL cells were treated with SAHA and PEITC under different
treatment conditions. To inhibit cystine uptake by the stromal
cells, (S)-4-CPG was added at the beginning of the stromal cell
seeding. To inhibit cystine uptake by the stromal cells, the CLL
and HS5 cells in co-culture were incubated with S-4-CPG (500
µM) for 24 h, and then exposed to SAHA (2 µM) for 48
h. Cell viability was determined using flow cytometry (FACSCalibur;
BD Biosciences), following double-staining with Annexin V-FITC and
PI. All assays were repeated at least three times using primary CLL
cells from different patient samples.
Analysis of cellular GSH levels
GSH was quantified using a GSH assay kit, based on
enzymatic recycling reactions. Following CLL cell culture under
various experimental conditions, the cells were collected,
sonicated at speed four (five times) at 4°C, and de-proteinated by
precipitation with an equal volume of 10% metaphosphoric acid
(Sigma-Aldrich). The precipitated proteins were removed by
centrifugation at 3,000 × g for 5 min at 4°C. The supernatant was
collected, neutralized with triethanolamine (cat. no. T58300;
Sigma-Aldrich) and assayed for GSH (reduced) and GSSG (oxidized)
using a Cayman GSH assay kit, according to the manufacturer's
instructions. The data were obtained from triplicate
measurements.
Detection of cellular ROS levels
The cellular ROS levels were measured by incubating
the CLL cells (1×106 cells) with 1 µM
CM-H2DCF-DA for 60 min at 37°C in the dark, followed by
analysis using flow cytometry, as previously described (8). The ROS levels in the viable cells
were determined with a chemical probe (CM-H2DCF-DA),
using forward scatter/side scatter gating to differentiate dead and
viable cells, as previously described (8).
Analysis of mitochondrial oxidative
damage, cytochrome c release, caspase-3 activation and
mitochondrial transmembrane potential
Cardiolipin is the predominant lipid component of
the internal mitochondrial membrane, and contributes to the
maintenance of its structure (21). The fluorescent dye, NAO, which
specifically binds to cardiolipin, is used to measure the oxidation
of cardiolipin (22). Following
CLL cell culture in the different conditions, the cells were
treated with or without SAHA, and the samples were labeled with 50
nM NAO for 15 min and analyzed using flow cytometry, as previously
described (23). A cytochrome
c release kit (EMD Millipore, San Diego, CA, USA) and
caspase-3 activation assay kit were used to measure the loss of
mitochondrial cytochrome c and the levels of activated
caspase-3, according to the manufacturer's instructions.
Rhodamine-123 (Invitrogen Life Technologies) was used to evaluate
mitochondrial transmembrane potential. The CLL cells were labeled
with 1 µM Rhodamine-123 for 60 min and analyzed using flow
cytometry, as previously described (24).
Western blot analysis
The CLL cells were solubilized in buffer containing
10 mM Tris-Hcl (pH 7.6; car. no. T1503; Sigma-Aldrich), 1% SDS
(cat. no. L3771; Sigma-Aldrich) and protease inhibitor (cat. no.
11836170001; Roche Diagnostics; Pleasanton, CA, USA). The proteins
were quantified using a bicinchoninic acid assay protein assay kit
(cat. no. 23225; Pierce Biotechnology, Rockford, IL, USA), and then
adjusted to 2 µg/ml with sample buffer containing 250 mM
Tris-Hcl (pH 6.8; cat. no. T1503; Sigma-Aldrich), 4% SDS (cat. no.
L3771; Sigma-Aldrich), 10% glycerol (cat. no. G5516;
Sigma-Aldrich), 0.006% bromophenol blue (cat. no. B0126;
Sigma-Aldrich) and 2% mercaptoetanol (cat. no. M6250;
Sigma-Aldrich). The cell lysates were heated at 95°C for 10 min,
and equal quantities of protein were electrophoresed on SDS-PAGE in
a Mini-Protean II Dual Slab Cell (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). The proteins were then transferred onto
nitrocellulose membranes using a Mini Trans-Blot Transfer Cell
(Bio-Rad Laboratories, Inc.). The transfer was performed at 4°C for
2 h at a constant voltage setting of 110 V. The blots were blocked
in 5% nonfat milk for 1 h at room temperature. The membranes were
then probed with the following primary antibodies: Rabbit
anti-human GCLC polyclonal antibody (cat. no. sc-28965, Santa Cruz
Biotechnology, Inc.) at a 1:1,000 dilution; rabbit anti-human Nrf2
polyclonal antibody cat. no. sc-13032; Santa Cruz Biotechnology,
Inc.) at a 1:1,000 dilution; rabbit anti-human Mcl1 polyclonal
antibody (cat. no. sc-819; Santa Cruz Biotechnology, Inc.) at a
1:1,000 dilution;and mouse anti-human actin monoclonal antibody
(cat. no. MA5-11869; Pierce Biotechnology) at a 1:10,000 dilution.
After 2 h incubation at room temperature, the blots were washed
three times for 10 min in PBS containing 0.1% Tween-20 (cat. no.
P1379; Sigma-Aldrich), and then incubated for 1 h at room
temperature in the following secondary antibodies: Goat anti-rabbit
polyclonal antibody (cat. no. 31210; Pierce Biotechnology) for
GCLC, Nrf2 and Mcl1 detection at a 1:3,000 dilution; and goat
anti-mouse polyclonal antibody (cat. no. 62-6700; Pierce
Biotechnology) for actin detection at a 1:20,000 dilution. The
blots were then washed three times for 10 min in the same buffer as
above, incubated in enhanced chemiluminescence detection reagents
(GE Healthcare Life Sciences, Chalfont, UK) for 1 min. The blots
were then exposed to an X-OMAT AR x-ray film (Kodak, Rochester, NY,
USA) for between 10 sec and 5 min.
Immunofluorescence and confocal
microscopy
The CLL cells were cytospun at 20 × g for 5 min at
room temperature, fixed with 3.7% (v/v) paraformaldehyde,
permeabilized with 0.2% Triton X-100 and blocked with 5% (w/v) BSA.
The fixed CLL cells were incubated with rabbit anti-human
polyclonal Nrf2 antibody (1:50; cat no. sc-13032; Santa Cruz
Biotechnology, Inc.), at 4°C overnight, followed by incubation with
Alexa-Fluor-594 goat anti-rabbit polyclonal antibody (1:400; cat.
no. A11594; Molecular Probes at room temperature for 1 h. Finally,
the slides were washed with phosphate-buffered saline, mounted and
counterstained with mounting medium supplemented with DAPI prior to
examination with a Nikon Eclipse TE2000 confocal microscope and
analysis with Nikon EZ-C1 3.80 software (Nikon Corporation, Tokyo,
Japan).
Determination of the effect of oxygen
levels on CLL cell viability
HS5 cells (5×104 cells/ml) were seeded in
24-well plates and allowed to adhere and grow overnight prior to
the addition of CLL cells. The CLL cells were isolated from blood
samples and incubated overnight at 37°C with RPMI 1640 medium
supplemented with 10% fetal bovine serum, penicillin (100 U/ml) and
streptomycin (100 µg/ml). Subsequently 1×106 CLL
cells were transferred into 24-well plates with or without a
pre-seeded stromal cell layer at a density of 5×104
cells per well, and incubated in normoxic or hypoxic conditions (5
and 2%, respectively) for 1 day at 37°C. The CLL cells were then
treated with SAHA (2 µM) for 43 h, followed by PEITC (5
µM) for 5 h, at various oxygen levels, at 37°C. Cell
viability was determined using flow cytometry following
double-staining with Annexin V and PI. Hypoxic culture conditions
were created by incubating the cells in a sealed modular incubator
chamber flushed with 5% or 2% oxygen, 5% carbon dioxide and
balanced nitrogen.
Reverse transcription-quantitative polymerase chaIn
reaction (RT-qPCR) analysis. Total RNA was isolated from the cells
using an RNeasy kit (Qiagen, Valencia, CA, USA), followed by DNAase
(Ambion Life Technologies, Austin, TX, USA) treatment to remove any
contaminating DNA. The RNA was quantitated spectrophotometrically
at 260 nm (Du800 nucleic acid/protein analyzer; Beckman-Coulter,
Fullerton, CA, USA). Reverse transcription was performed using
TaqMan Reverse Transcription reagents (Applied Biosystems Life
Technologies, Foster City, CA, USA). Gene-specific primers for GCLC
(forward 5′-AGAGAAGGGGAAAGGACAA-3′ and reverse
5′-GTGAACCCAGGACAGCCTAA-3′) and β-actin (forward
5′-TCACCCACACTGTGCCCATCTACGA-3′ and reverse
5′-TGAGGTAGTCAGTCAGGTCCCG-3′) were used to amplify a segment of
reverse transcribed mRNA using an ABI 7700 sequence detection
system and SYBR reagents (Invitrogen Life Technologies). β-actin
was used as the internal control gene. Human gene-specific primers
were synthesized by Invitrogen Life Technologies. PCR reactions
were prepared with final concentrations of 1 µg cDNA and 200
nM primers. Thermal cycling conditions comprised a pre-heating
cycle for 10 min at 95°C for 50 cycles, then 95°C for 25 sec, 60°C
for 40 sec, and 70°C for 40 sec. Each measurement was performed in
triplicate, and the threshold cycle and the fractional cycle number
was determined. β-actin was used as the internal control gene.
Statistical analysis
All experiments were performed on CLL cells from at
least three patients, and stromal cells from three separate culture
flasks were used. Statistical significance was analyzed using
Student's t-test. P<0.05 was considered to indicate a
statistically significant difference. Bar graphs and plots were
generated using GraphPad Prism 5 software (GraphPad Software, Inc.,
La Jolla, CA, USA).
Results
Bone marrow stromal cells increase the
expression of GSH and decrease SAHA-mediated apoptosis in CLL
cells
Previous studies have demonstrated that HDACIs have
limited activity in patients with CLL (17,18).
Since the bone marrow is an important site for malignant cells, a
co-culture system using a bone marrow stromal cell line and primary
leukemia cells isolated from patients with CLL was used to
investigate the effect of bone marrow stromal cells and redox
change on the activity of SAHA in CLL cells. As shown in Fig. 1A, the HS5 bone marrow stromal cells
significantly reduced spontaneous CLL cell death. Incubation with 2
µM SAHA for 48 h induced 60–70% CLL cell death; however, HS5
cells markedly protected the CLL cells from SAHA-induced cell death
(50–80% viable cells; Fig.
1A).
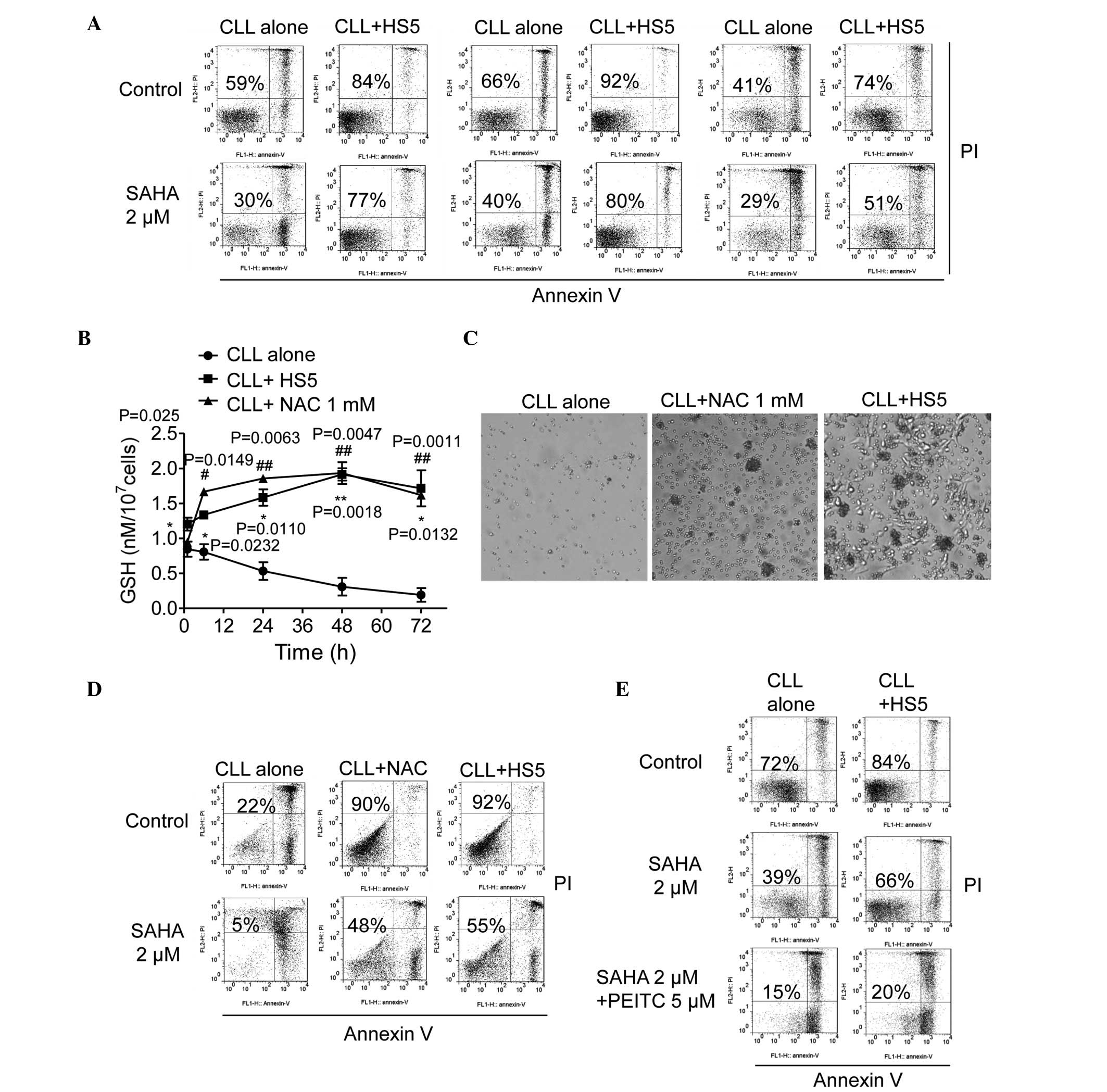 | Figure 1GSH-mediated stromal protection of
CLL cells from SAHA-induced cell death. (A) Protection of CLL cells
by HS5 cells in the presence and absence of SAHA. The CLL cells
were pre-cultured with HS5 cells for 24 h, followed by 2 µM
SAHA incubation for 48 h. Cell viability was measured using Annexin
V/PI double staining. Representative dot plots of a CLL sample are
shown, with numbers indicating the percentage of viable cells
(Annexin V/PI double negative). (B) GSH levels over time in CLL
cells cultured alone, with HS5 cells, or with NAC (1 mM). Values
are presented as the mean ± standard error of the mean of three
separate experiments using three CLL samples *P<0.05
and **P<0.01, CLL cells cultured alone, vs. CLL cells
cultured with HS5 cells; #P<0.05 and
##P<0.01, CLL cells cultured alone, vs. CLL cells
cultured with NAC. (C) NAC and HS5 cells promoted CLL cell survival
for 7 days. When stromal cells reached confluence, the CLL cells
were transferred to a new flask pre-seeded with HS5 cells. Images
were captured on day 7. (D) NAC and HS5 cells promoted CLL cell
survival in the presence and absence of SAHA. The CLL cells were
pre-cultured with HS5 cells or supplemented with NAC (1 mM) for 7
days, followed by 2 µM SAHA incubation for 48 h. Cell
viability was measured using Annexin V/PI double staining. The
number in each dot blot indicates the percentage of viable cells.
(E) Annexin V/PI assay of cell viability following CLL cell culture
alone or with HS5 cells, in the presence or absence of SAHA (2
µM; 48 h), or its combination with PEITC (5 µM; 5 h).
The number in each dot blot indicates the percentage of viable
cells. CLL, chronic lymphocytic leukemia; SAHA, suberoylanilide
hydroxamic acid; GSH, glutathione; PI, propidium iodide; PEITC,
β-phenylethyl isothiocyanate; NAC, N-acetylcysteine. |
The activity of HDACIs is affected by the redox
status of malignant cells (11,12).
GSH is the most abundant antioxidant buffer system, and may be
upregulated by stromal cells in the CLL cell (8). The present study subsequently
investigated the role of GSH in stromal-mediated protection against
SAHA action in the CLL cells. CLL cells cultured alone exhibited a
time-dependent decrease in levels of GSH, whereas the addition of
HS5 cells or supplementation with GSH precursor NAC maintained high
intracellular levels of GSH in the CLL cells (Fig. 1B). On day 3, the stromal cells and
NAC maintained the levels of GSH in the CLL cells in the range of
1.5–2 nmol/1×107 cells, whereas single-cultured CLL
cells without NAC supplementation had <0.5 nmol/1×107
cells (Fig. 1B). Similar to the
HS5 cells, supplementation of 1 mM NAC in the CLL cell culture
medium enhanced CLL cell viability for 7 days (Fig. 1C). The Annexin V/PI assay further
determined that CLL cells in the presence of HS5 cells or NAC were
90–92% viable, compared with the CLL cells cultured alone with low
levels of GSH, of which only 22% were viable following 7 days
incubation in vitro (Fig.
1D). Notably, incubation with 2 µM SAHA for a further 2
days caused extensive apoptosis in the CLL cells alone (95%),
whereas the NAC or HS5 cells conferred significant protection to
the CLL cells against SAHA, leading to 52 and 45% apoptotic cell
death, respectively (Fig. 1D).
Furthermore, the use of PEITC to rapidly deplete cellular GSH
inhibited stromal protection of the CLL cells against SAHA
toxicity, and enhanced the cytotoxic effects of SAHA in the
presence of stromal cells (Fig.
1E). These results suggested that the bone marrow stromal cells
protected the CLL cells from spontaneous and SAHA-induced
apoptosis, and GSH was critical in the stromal protection of CLL
cells against SAHA toxicity.
Bone marrow stromal cells reverse
SAHA-induced ROS damage to mitochondria
Concordant with previous reports of ROS induction by
HDACIs (11,25), SAHA induced ROS generation in the
CLL cells following 20 h of incubation (Fig. 2A). As CLL cells deplete GSH rapidly
and rely on stromal cells to maintain intracellular GSH levels
(8), the present study
hypothesized that GSH deletion and SAHA-induced ROS generation
cause a rapid redox imbalance and ROS-mediated mitochondrial
damage, whereas bone marrow stromal cells upregulate GSH and
reverse SAHA-induced damage. As shown in Fig. 2B, incubation of the CLL cells with
SAHA led to the rapid oxidation of cardiolipin, determined by the
loss of interaction with fluorescent NAO dye. The NAO fluorescent
signal increased between 18 and 80% following treatment with 2
µM SAHA at 22 h (Fig. 2B).
Notably, the HS5 cell or NAC-induced increase in intracellular
levels of GSH in the CLL cells largely reduced the SAHA-induced
loss of NAO signal between 80, and 28 and 29%, respectively
(Fig. 2B). A previous study
demonstrated that cardiolipin induces cytochrome c release
and caspase activation (21). As
shown in Fig. 2C and D, exposure
to 2 µM SAHA for 24 h induced mitochondrial cytochrome
c release and caspase 3 activation. The HS5 cells and NAC
also prevented SAHA-induced cytochrome c release, and
reduced caspase activation between 38, and 3 and 6%, respectively
(Fig. 2C and D). These results
suggested that bone marrow stromal cells, which increased the
levels of intracellullar GSH in the CLL cells, reversed
SAHA-induced ROS damage to the mitochondria in a similar manner to
that by the NAC GSH precursor.
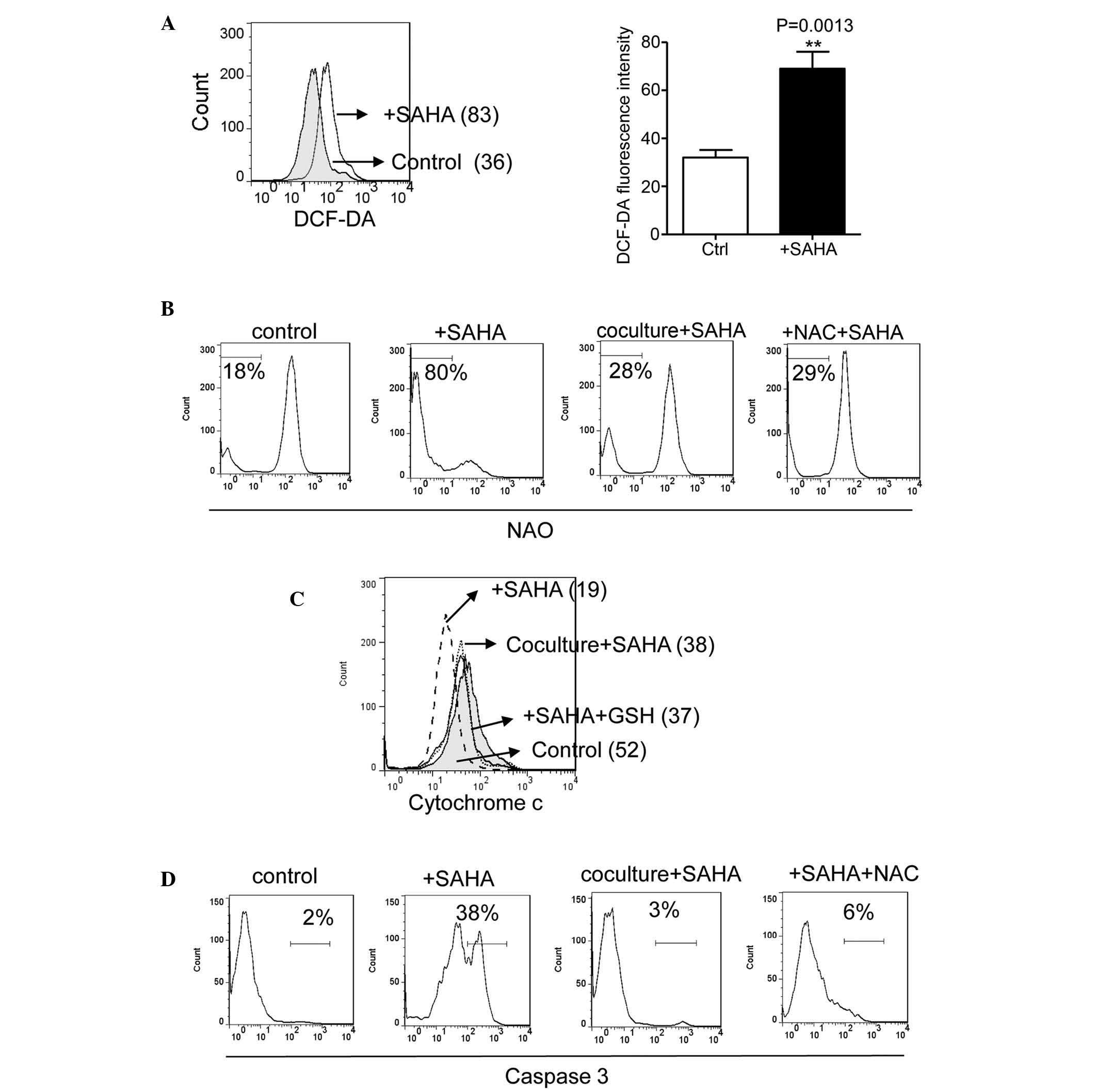 | Figure 2Bone marrow stromal cells reverse
SAHA-induced ROS damage to mitochondria. (A) Determination of
cellular ROS in CLL cells with or without SAHA (2 µM, 20 h)
incubation, determined using flow cytometric analysis. A
representative plot (left, mean value of relative intensity
indicated) and quantitative comparison of the mean values ±
standard deviation from five CLL samples are shown (right,
**P<0.01, vs. control). (B) Change of cardiolipin
oxidation in CLL cells treated with SAHA at the indicated
conditions. CLL cells were pre-cultured with HS5 cells or
supplemented with NAC (1 mM) for 24 h, followed by 2 µM SAHA
incubation for 22 h. Cardiolipin oxidation was measured by flow
cytometry using NAO staining. Representative histograms are shown.
The numbers indicate the gating of the subpopulation of CLL cells
exhibiting loss of cardiolipin signal due to oxidation. (C)
Determination of mitochondrial cytochrome c in CLL cells
pre-cultured with HS5 cells or supplemented with NAC (1 mM) for 24
h, followed by 2 µM SAHA incubation for 24 h. The overlays
show the distribution of mitochondrial cytochrome c
fluorescence intensity of each cell population, with the mean value
of the relative intensity indicated. Representative histograms of a
CLL patient sample are shown. (D) Caspase 3 activation in CLL cells
pre-cultured with HS5 cells or supplemented with NAC (1 mM) for 24
h, followed by 2 µM SAHA incubation for 24 h. The numbers
indicate the gating of a subpopulation of cells with positive
caspase 3 activation. Representative histograms of three separate
experiments are shown. CLL, chronic lymphocytic leukemia; SAHA,
suberoylanilide hydroxamic acid; ROS, reactive oxygen species; NAC,
N-acetylcysteine. |
SAHA increases GSH synthesis, potentiates
stromal-mediated GSH upregulation in CLL cells and protects cells
from SAHA-mediated ROS damage and apoptosis
A previous study suggested that the antioxidant
system emerged to protect leukemia cells during cellular selection
with HDACI (12). The present
study investigated whether SAHA-induced ROS generation in CLL cells
stimulated the compensatory antioxidant system. The NF-E2-related
factor 2 (Nrf2) antioxidant response signaling pathway is the
primary cellular defense mechanism against the cytotoxic effects of
oxidative stress (22). As shown
in Fig. 3A, incubation with 2
µM SAHA for 20 h upregulated the expression of Nrf2 in the
CLL cells. It has been suggested that Nrf2 localizes to the
cytoplasm to interact with Kelch-like ECH-associating protein 1
(Keap1) in the normal physiological state (26). The activity of the Nrf2 signaling
system is dependent upon redox homeostasis in the cells, and
modification of Keap1 by ROS leads to Nrf2 dissociation and
translocation to the nucleus, leading to transactivation of its
downstream target genes (27). The
present study used immunofluorescence staining with confocal
microscopy to identify the subcellular location of Nrf2
translocation. Nrf2, revealed using Alexa Fluor® 594 (in
red) induced a condensed, ring-like staining pattern in the
cytoplasm around the nucleus in untreated CLL cells (Fig. 3B). However, treatment with 2
µM SAHA for 20 h resulted in an accumulation of Nrf2 within
the nucleus, which was stained by DAPI, a DNA binding dye (Fig. 3B). These results suggested that
SAHA induced Nrf2 translocation between the cytoplasm and nucleus
in the CLL cells.
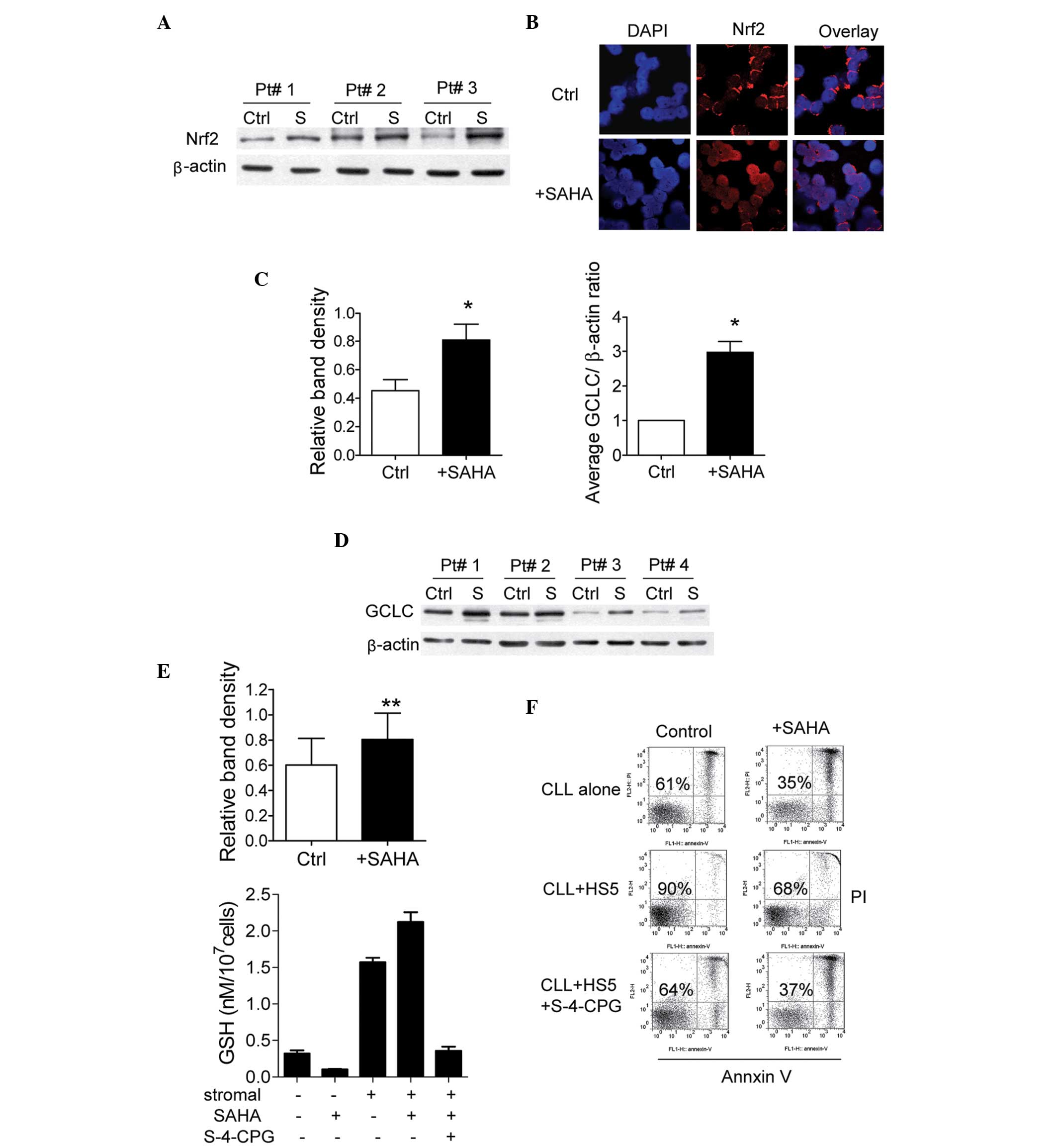 | Figure 3Effect of SAHA on levels of
GSH-associated enzyme and GSH in CLL cells co-cultured with stromal
cells. (A) SAHA increased the expression of Nrf2 in CLL cells. CLL
cells were treated with 2 µM SAHA for 20 h, and cell lysates
were assayed for Nrf2 using western blot analysis. Representative
western blot results from three samples from patients with CLL are
shown. The right panel shows the quantification of Nrf2 band
density of eight CLL samples, with β-actin expression as an
internal control (mean ± standard deviation; *P<0.05;
Ctrl, control cells without treatment; S, SAHA treatment). (B) SAHA
induced the translocation of Nrf2 between the cytosol and nucleus.
SAHA (2 µM) was added to the CLL cells for 20 h, and the
cells were cytospun and immunostained with Nrf2 antibodies, and
observed using a confocal laser scanning microscope. The nuclei
were stained with 4,6-diamidino-2-phenylindole. (C) Upregulation of
mRNA expression of GCLC following SAHA treatment. CLL cells were
treated with 2 µM SAHA for 22 h and the GCLC mRNA expression
was examined using reverse transcription-quantitative polymerase
chain reaction. (D) SAHA increases the expression of GCLC in CLL
cells. CLL cells were treated with 2 µM SAHA for 24 h, and
the cell lysates were then assayed for the expression levels of
GCLC by western blot analysis. The upper panel shows the
representative western blot results from samples of four patients
with CLL. The lower panel shows the quantification of GCLC band
density of eight CLL samples, with β-actin as the internal control
(mean ± standard deviation; **P<0.01. (E) Treatment
with SAHA enhanced stromal-mediated GSH upregulation in CLL cells.
The CLL cells were treated with 2 µM SAHA for 48 h in the
presence or absence of HS5 cells. In another treatment group, CLL
and HS5 cells in co-culture were incubated with cystine transporter
inhibitor S-4-CPG (500 µM) for 24 h, then exposed to SAHA (2
µM) for 48 h. Values are presented as the mean ± standard
deviation of three independent experiments using three CLL samples.
(F) Sensitization of CLL cells to SAHA by inhibiting the cystine
transporter with S-4-CPG. CLL and HS5 cells in co-culture were
incubated with S-4-CPG (500 µM) for 24 h, then exposed to
SAHA (2 µM) for 48 h. Cell viability was analyzed using an
Annexin V/PI assay. Representative dot plots are shown. CLL,
chronic lymphocytic leukemia; SAHA, suberoylanilide hydroxamic
acid; GSH, glutathione; PI, propidium iodide; Nrf2, nuclear
factor-E2-related factor 2; CPG, carboxyphenylglycine Ctrl,
untreated control; S, SAHA treatment. |
The expression of GCLC, a rate-limiting enzyme in
GSH synthesis, is controlled by the Nrf2 transcription factor
(28). In the present study, the
mRNA expression levels of GCLC were examined. As shown in Fig. 3C, GCLC gene expression was
upregulated in CLL cells following treatment with 2 µM SAHA
for 22 h. The protein expression levels of GCLC also increased
following treatment with SAHA (Fig.
3D). As determined by measurement of cellular GSH levels, SAHA
treatment decreased levels of GSH in the CLL cells (Fig. 3E). These results suggested that the
increase in GSH enzyme synthesis induced by SAHA treatment failed
to promote GSH synthesis in the CLL cells, which was entirely
distinct from the results in other types of leukemia (12). However, in the presence of HS5
cells, SAHA treatment increased, rather than decreased, levels of
GSH in the CLL cells (Fig. 3E). It
has been suggested that GSH synthesis is limited by the cellular
levels of its cysteine substrate (28). Our previous report demonstrated
that bone marrow stromal cells effectively imported cystine by the
Xc-transporter, and converted it to cysteine to supply CLL cells
for GSH synthesis (8). As shown in
Fig. 3E, the Xc-transporter
inhibitor, S-4-CPG, prevented SAHA-mediated GSH upregulation in the
co-cultured CLL cells. In addition, sub-toxic concentration levels
of S-4-CPG (500 µM) inhibited stromal cell protection and
enhanced SAHA-induced cytotoxicity, to levels comparable to those
observed in CLL cells without stromal protection (Fig. 3F). These results suggested that
stromal-produced GSH substrate and SAHA-induced GSH synthesis
enzyme may promote an increase in GSH in CLL cells, and protect
cells from SAHA-mediated ROS damage and apoptosis.
Treatment with PEITC overcomes stromal
protection of CLL cells against SAHA toxicity by inhibiting stromal
and SAHA-mediated upregulation of GSH in CLL cells
PEITC is a compound known to conjugate with GSH,
leading to the expo-ration and depletion of cellular GSH (23), which is considered to be an
important mechanism underlying PEITC-induced ROS stress in cancer
cells. As shown in Fig. 4A, the
HS5 cells upregulated the levels of GSH in the CLL cells, and
treatment with 2 µM SAHA caused a further increase in GSH
following 48 h of co-culture in the CLL cells. However, the
addition of 5 µM PEITC for 5 h markedly decreased levels of
GSH in the CLL cells. Cellular ROS levels in the CLL cells were
then examined under various conditions. In Fig. 4B, flow cytometric analysis
demonstrated that both 2 µM SAHA and 5 µM PEITC
increased ROS levels in the CLL cells co-cultured with the HS5
cells. The combination of these two compounds resulted in the
additive accumulation of ROS, which was reversed by pre-treatment
with 1 mM NAC (Fig. 4B and C).
These data suggested that PEITC depleted stromal cell-upregulated
GSH in the CLL cells, and mediated SAHA induced-ROS generation in
the co-cultured CLL cells.
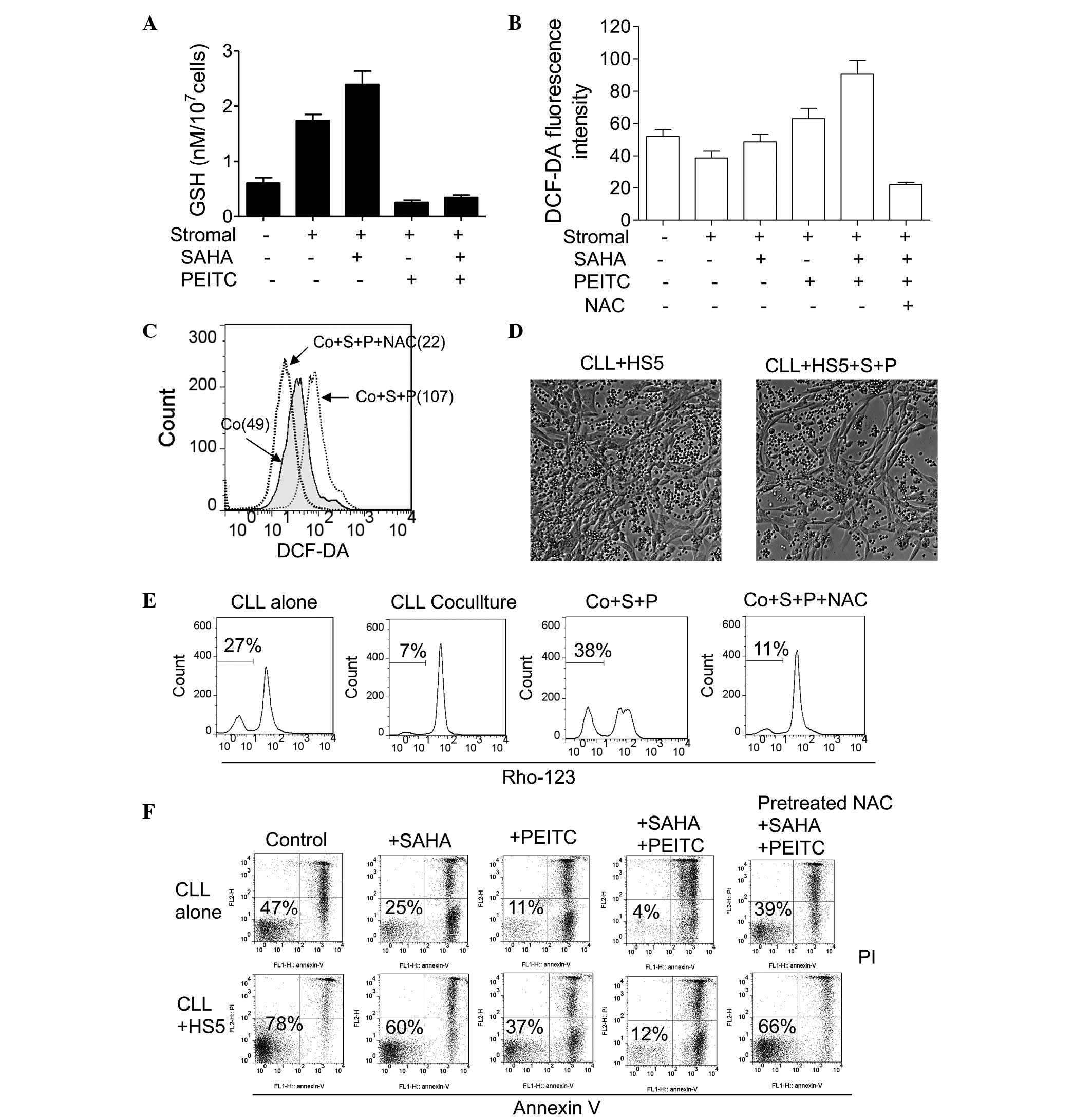 | Figure 4Treatment combination of PEITC and
SAHA inhibits GSH upregulation in CLL cells, and induces
ROS-mediated apoptosis. (A) Comparison of GSH levels in CLL cells
treated with SAHA (2 µM; 48 h) or/and PEITC (5 µM; 5
h) in the presence of HS5 cells. The values are presented as the
mean ± standard deviation of three separate experiments using four
CLL samples. (B) Determination of ROS levels in CLL cells treated
with SAHA (2 µM; 48 h) or/and PEITC (5 µM; 5 h) in
the presence of HS5 cells, determined using flow cytometric
analysis. Quantitative comparison of mean values ± standard
deviation from four CLL samples are shown. (C) Pre-treatment with
NAC (1 mM) blocked the upregulation of ROS induced by the
combination of SAHA (2 µM; 48 h) and PEITC (5 µM; 5
h). The overlays show the distribution of ROS fluorescence
intensity of each cell population, with the mean value of the
relative intensity indicated. Representative histograms of a CLL
patient sample are shown. (D) Combination of SAHA (2 µM; 48
h) and PEITC (5 µM; 5 h) selectively eliminated CLL cells,
sparing HS5 cells. Images of the cells were captured using a Nikon
Eclipse TE2000 microscope. (E) Determination of mitochondrial
transmembrane potential in CLL cells pre-cultured with HS5 cells or
supplemented with NAC (1 mM) for 24 h followed by the
co-administration of SAHA and PEITC. The numbers indicate the
gating of the subpopulation of cells with loss of transmembrane
potential. (F) Apoptosis induced by SAHA (2 µM; 48 h) or/and
PEITC (5 µM; 5 h) with or without HS5 cell co-culture was
inhibited by pre-treatment with NAC (1 mM). Representative dot
plots of a CLL sample are shown; the numbers indicate the
percentage of viable cells (Annexin V/PI double negative). CLL,
chronic lymphocytic leukemia; SAHA, suberoylanilide hydroxamic
acid; GSH, glutathione; PEITC, β-phenylethyl isothiocyanate; NAC,
N-acetylcysteine; Co, co-culture; S, SAHA treatment; P, PEITC
treatment; ROS, reactive oxygen species; PI, propidium iodide. |
Based on the above results, the present study
hypothesized that PEITC may circumvent stromal protection against
SAHA-induced ROS attack, and maximize its antileukemic activity by
depleting stromal cell-upregulated cellular GSH in CLL cells. As
shown in Fig. 4E, CLL cells alone
exhibit 27% loss of transmemebrane potential due to endogenous
oxidative stress and spontaneous apoptosis, whereas HS5 stromal
cells decreased CLL cell transmembrane potential loss to 7%.
Treatment with 2 µM SAHA for 22 h followed by 5 µM
PEITC treatment for 5 h resulted in a 38% loss of transmembrane
potential, indicating early induction of apoptosis. Notably, the
effects of the combination were reversed by pre-teatment with 1 mM
NAC (Fig. 4E), indicating that
accumulation of ROS in the CLL cells contributed to the cytotoxic
effect induced by the combination of SAHA and PEITC.
The effect of the combination of SAHA and PEITC was
also examined using an Annexin V/PI assay. As shown in Fig. 4F, incubation with 2 µM SAHA
for 48 h or 5 µM PEITC for 5 h was toxic to single-cultured
CLL cells, and HS5 cells protected the CLL cells from the cytotoxic
effects of SAHA and PEITC. However, treatment with 2 µM SAHA
for 43 h followed by 5 µM PEITC treatment for 5 h eliminated
>85% of the CLL cells with or without stromal protection.
Concordantly, treatment with NAC largely inhibited the combination
effects of SAHA and PEITC (Fig.
4F), which further confirmed that SAHA and PEITC combination
led to ROS-mediated cell death. Furthermore, the combination of the
two drugs eliminated the CLL cells cultured with HS5 cells,
although the HS5 stromal layers were not visibly affected (Fig. 4D), indicating that the combination
of SAHA and PEITC preferentially destroyed the CLL cells
co-cultured with stromal cells, but spared the normal stromal
cells.
PEITC induces rapid degradation of
stromal cell and SAHA treatment-upregulated Mcl1 in CLL cells
Anti-apoptotic proteins, including B-cell lymphoma
(Bcl)-2 and Mcl1 are important in cancer cell survival. Unlike
Bcl-2, Mcl1 has a short half-life and its expression is tightly
regulated by survival and apoptotic signals (29). In CLL cells, Mcl1 is particularly
important in microenvironment-mediated survival and drug resistance
(30). Treatment with 5 µM
SAHA for 48 h caused an increase in the mRNA expression of Mcl1 in
the CLL cells (Fig. 5A). The
protein expression levels of Mcl1 in the CLL cells were also
increased following treatment with SAHA (Fig. 5B), indicating that SAHA upregulated
the expression of Mcl1 in the CLL cells. In addition, co-culture
with HS5 cells or treatment with NAC significantly increased the
levels of Mcl1 in the CLL cells (Fig.
5C). Mcl1 has been demonstrated to be stabilized by GSH via
glutathionylation (23). It is
likely that stromal cells stabilize Mcl1 in CLL cells by
upregulating GSH levels, thereby promoting glutathionylation of
Mcl1. When the CLL cells co-cultured with HS5 cells or pre-treated
with NAC were incubated with 5 µM SAHA for 48 h, a 3-fold
increase in the levels of Mcl1 were observed, compared with the CLL
cells cultured alone or without NAC (Fig. 5C). These data suggested that the
levels of Mcl1 in the CLL cells were increased in the presence of
bone marrow stromal cells, and were further enhanced by SAHA
treatment.
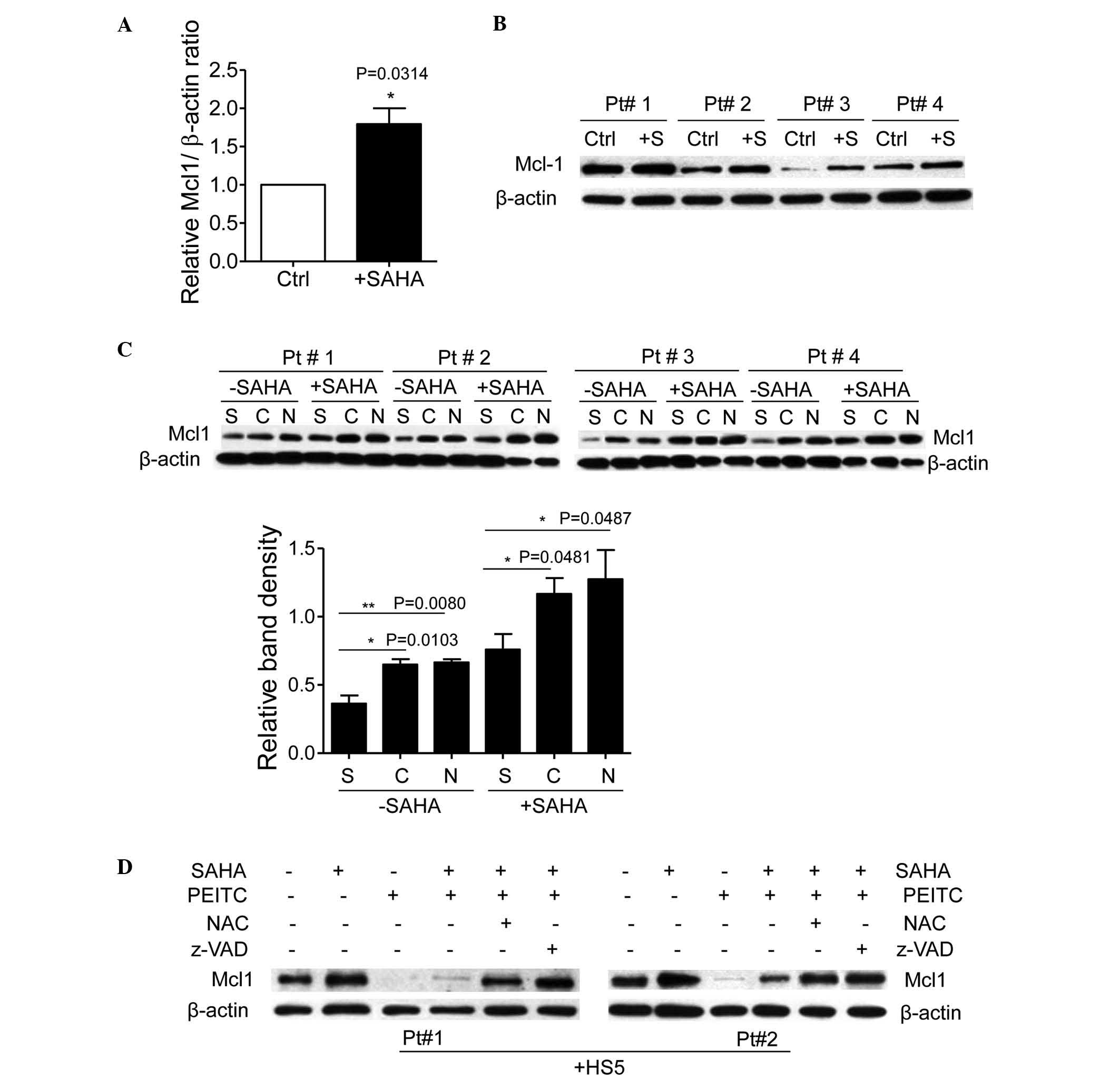 | Figure 5PEITC induces degradation of stromal
cell and SAHA treatment-upregulated Mcl1 in CLL cells. (A)
Upregulation of the mRNA expression levels of Mcl1 following SAHA
treatment. CLL cells were treated with 2 µM SAHA for 48 h
and mRNA expression levels of Mcl1 were examined using reverse
transcription-quantitative polymerase chain reaction analysis. (B)
SAHA increased the protein expression of Mcl1 in CLL cells. CLL
cells were treated with 2 µM SAHA for 48 h, and cell lysates
were then assayed for expression of Mcl1 using western blot
analysis. The panel shows the representative western blots of four
samples of patients with CLL. (C) Expression levels of Mcl1 in CLL
cells co-cultured with HS5 cells or supplemented with 1 mM NAC with
or without SAHA treatment (2 µM; 48 h). The upper panel
shows the representative western blots of four samples of patients
with CLL. The lower panel shows the quantification of Mcl1 band
density, using β-actin as an internal control (mean ± standard
deviation). (D) PEITC (5 µM; 5 h) decreased expression
levels of Mcl1 in CLL cells co-cultured with HS5 cells with or
without SAHA treatment. NAC or caspase inhibitor (Z-VAD-fmk)
suppressed PEITC-induced Mcl1 degradation. CLL cells were
pretreated with 1 mM NAC for 24 h or 20 µM Z-VAD-fmk for 30
min prior to treatment with PEITC. A representative western blot
from experiments with two CLL samples is shown. CLL, chronic
lymphocytic leukemia; SAHA, suberoylanilide hydroxamic acid; PEITC,
β-phenylethyl isothiocyanate; NAC, N-acetylcysteine; Mcl1, myeloid
cell leukemia 1; Ctrl, untreated control cells; +S, SAHA treatment;
S, single culture of CLL cells alone; C, co-cultured with stromal
cells; N, supplemented with NAC. |
Due to the fact that PEITC depletes GSH and causes
protein deglutathionylation, including Mcl1 (23), the present study hypothesized that
PEITC may cause degradation of Mcl1 and circumvent stromal cell and
SAHA-mediated Mcl1 upregulation in CLL cells. To investigate this
possibility, the expression of Mcl1 was examined in CLL cells
treated with the combination of SAHA and PEITC in the presence of
HS5 cells. As shown in Fig. 5D,
SAHA incubation increased the expression of Mcl1 in the CLL cells
co-cultured with HS5 cells, however, PEITC substantially abrogated
the expression of Mcl1 upregulated by SAHA treatment in the CLL
cells. Treatment with NAC to replenish GSH levels significantly
prevented the PEITC-mediated decrease in the levels of Mcl1.
Notably, the caspase inhibitor, Z-VAD.fmk, inhibited PEITC-induced
Mcl1 degradation (Fig. 5D),
indicating that caspase was the major protease cleaved by Mcl1
following treatment with PEITC. These results suggested that
stromal cells and SAHA treatment upregulated the expression of Mcl1
in the CLL cells, however, PEITC induced deglutathionylation of
Mcl1 followed by caspase cleavage and degradation, which likely
potentiated HDACI-mediated apoptosis in the CLL cells co-cultured
with the stromal cells.
Combination treatment with SAHA and PEITC
effectively eliminates CLL cells in the presence of bone marrow
stromal cells
Annexin V/PI staining was used to examine the
effects of SAHA, PEITC, and their combination on primary CLL cells
from multiple patients cultured on stromal layers. Fresh CLL cells
isolated from 10 patients with CLL were co-cultured with HS5 cells
and treated with 2 µM SAHA alone for 48 h, 5 µM PEITC
alone for 5 h, or the combination of 2 µM SAHA for 43 h
followed by 5 µM PEITC for 5 h, respectively. As shown in
Table I, SAHA alone caused 20–60%
cell death in the co-cultured CLL cells isolated from different
patient samples. PEITC was more toxic towards the CLL cells and
caused 40–70% cell death in these samples. Notably, the combination
of SAHA and PEITC caused >80% loss of viability in the
co-cultured CLL cells. These results indicated that the effect of
the combination was more than an additive effect in all 10 patient
samples, and the combination of PEITC and SAHA effectively
destroyed the CLL cells co-cultured with the stromal cells.
 | Table ICytotoxicity of SAHA, PEITC and their
combination in CLL cells co-cultured with HS5 cells. |
Table I
Cytotoxicity of SAHA, PEITC and their
combination in CLL cells co-cultured with HS5 cells.
| Patient | Residual viable
cells (%)
| Combination
effect |
|---|
| 2 µM
SAHA | 5 µM
PEITC | 2 µM SAHA +
5 µM PEITC
|
|---|
| Observed | Predicted |
|---|
| 1 | 0.6 | 0.37 | 0.10 | 0.22 | >additive |
| 2 | 0.54 | 0.53 | 0.08 | 0.29 | >additive |
| 3 | 0.78 | 0.45 | 0.13 | 0.35 | >additive |
| 4 | 0.41 | 0.27 | 0.05 | 0.11 | >additive |
| 5 | 0.81 | 0.39 | 0.21 | 0.32 | >additive |
| 6 | 0.65 | 0.31 | 0.17 | 0.20 | >additive |
| 7 | 0.72 | 0.35 | 0.12 | 0.25 | >additive |
| 8 | 0.59 | 0.29 | 0.07 | 0.17 | >additive |
| 9 | 0.66 | 0.32 | 0.11 | 0.21 | >additive |
| 10 | 0.71 | 0.40 | 0.19 | 0.28 | >additive |
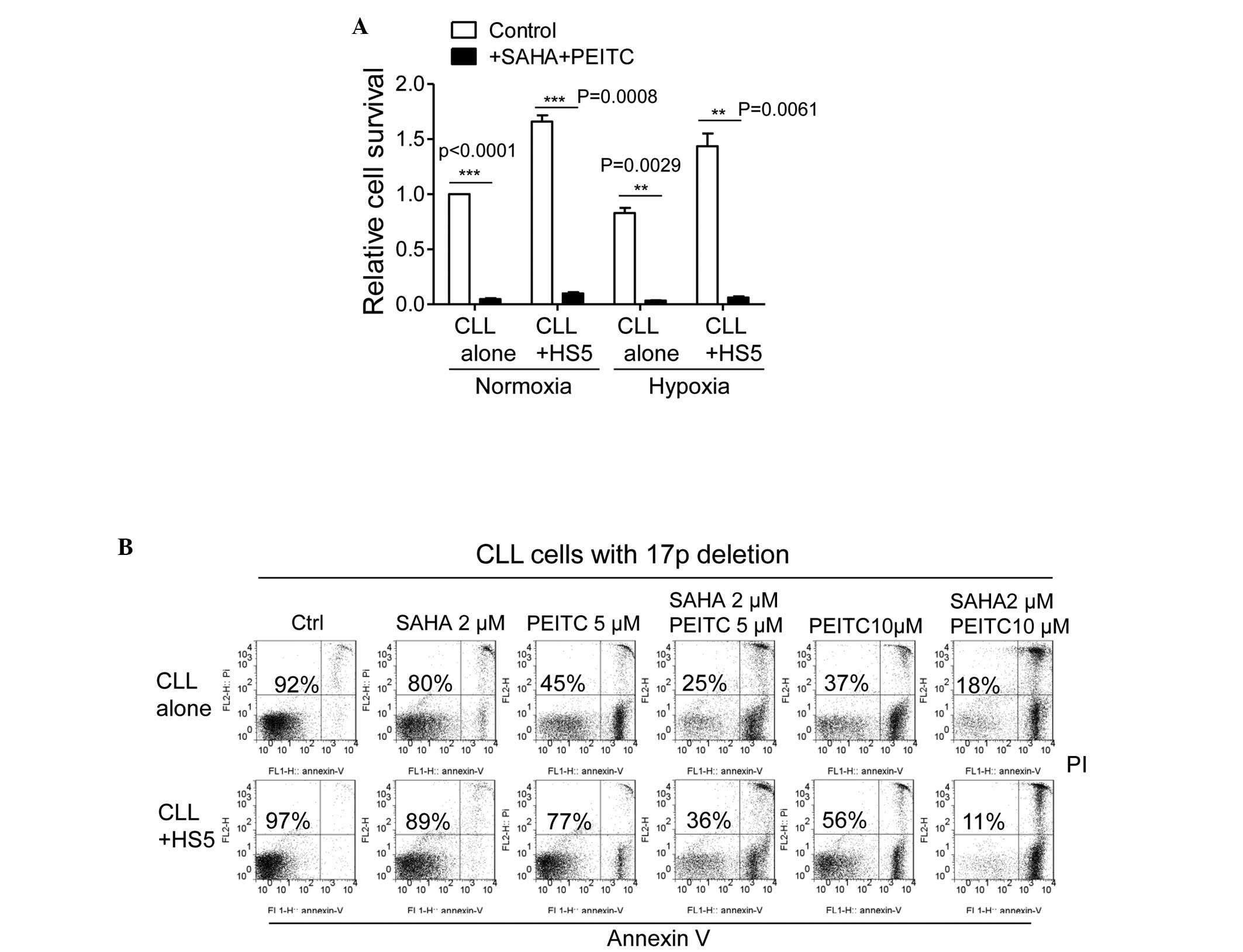
Due to the hypoxic bone marrow environment, the
effects of treatment combination of SAHA and PEITC in CLL cells
cultured on a stromal layer in various oxygen condition was
examined. As shown in Fig. 6A,
bone marrow stromal cells enhanced CLL viability under ambient or
hypoxic conditions. Concordantly, significant apoptotic cell death
induced by the combination of 2 µM SAHA and 5 µM
PEITC in the CLL cells was observed under ambient oxygen and
hypoxic conditions, suggesting that co-administration of SAHA and
PEITC may effectively eliminate CLL cells in the hypoxic bone
marrow environment.
Clinical studies have demonstrated that the loss of
p53 in CLL cells due to a chromosome 17p deletion is known to cause
drug resistance and poor prognosis in patients with CLL (1,23,31).
As shown in Fig. 6B, primary CLL
cells isolated from a patient with 17p deletion were highly
resistant to SAHA treatment in the presence or absence of stromal
cells. Compared with p53-positive CLL cells, which exhibited 30–60%
viability following PEITC treatment in the presence of HS5 cells
(Table I), the p53-negative CLL
cells co-cultured with HS5 cells exhibited 77% viability following
treatment with 5 µM PEITC for 5 h (Fig. 6B). The combination of 2 µM
SAHA and 5 µM PEITC resulted in a higher than additive
effect on cell death with or without HS5 cells. Notably, 10
µM PEITC was able to eliminate ~50% of the p53-negative CLL
cells in the presence of HS5 cells, and was highly effective when
combined with 2 µM SAHA, resulting in 90% cell death
(Fig. 6B). These data suggested
that the combination of PEITC and SAHA was highly effective at
eliminating CLL cells containing the 17p deletion in the presence
of bone marrow stromal cells.
Discussion
CLL is characterized by the abnormal accumulation of
functionally defective B lymphocytes in the blood, bone marrow,
spleen and other organs, eventually leading to organ failure and
ultimately the patient succumbing to the disease (3,32).
Cross-talk between CLL cells and bone marrow stromal cells favors
CLL progression and drug resistance (8). Therefore, disrupting the interaction
between CLL cells and their environment is an attractive, novel
strategy for treating patients with CLL. Our previous study
reported an important metabolic interaction between CLL cells and
bone marrow stromal cells, which increased GSH synthesis and thus
increased the ability of CLL cells to maintain the redox balance,
promoting cell survival and drug resistance (8). It was demonstrated that the GSH
depleting agent, PEITC, was effective at inhibiting stromal
protection in CLL cells and enhancing drug sensitivity (10). The aims of the present study were
to further evaluate this biochemical intervention strategy in CLL
cells, and to develop a combined treatment strategy to overcome
drug resistance and eliminate residual CLL cells.
HDACIs are a novel class of targeted anticancer
agents (33). HDACIs have multiple
biological effects, including histone and non-histone protein
acetylation, apoptosis, cell cycle arrest and senescence (10). A previous study reported that
HDACIs generate ROS in cancer cells (12). Multiple preclinical studies and
clinical data support the use of HDACI in combination with other
drugs for the treatment of various types of cancers (12,34,35).
SAHA is a small molecule inhibitor of class I and II HDACs, and
preclinical data has suggested a role for SAHA as a potential novel
treatment in several tumor types, including hematological
malignancies (36,37). SAHA has been approved for use
against cutaneous T cell lymphoma (38). However, the activity of SAHA has
been modest in patients with leukemia (39). In patients with CLL, initial
monotherapy clinical trials with various HDACIs revealed limited
activity of the drug (17,18). The present study demonstrated that
bone marrow stromal cells protected CLL cells against SAHA
cytotoxicity, indicating that bone marrow stromal cells may affect
SAHA sensitivity in patients with leukemia. Notably, combined
treatment with SAHA and PEITC induced extensive apoptosis in the
CLL cells in the presence of stromal cells. Flow cytometric
analysis following Annexin V/PI staining revealed that the effect
of the combination of drugs was more than additive in the CLL cells
from multiple patient samples co-cultured with stromal cells. These
results indicated that a greater level of efficacy was achieved
with combined treatment of SAHA and PEITC.
A previous study suggested that ROS are important in
the action of SAHA in acute myeloid leukemia cells, and that the
cellular redox status regulates sensitivity to SAHA treatment
(12). Previous studies using
primary leukemia cells isolated from the blood samples of patients
with CLL demonstrated that CLL cells are intrinsically under high
oxidative stress, compared with normal lymphocytes (7), and are highly sensitive to agents
that cause further ROS stress (7,23).
Therefore, CLL cells may have increased dependency on the GSH
antioxidant system. The results of the present study suggested that
single-cultured CLL cells exhibited low intracellular levels of
GSH, and were sensitive to SAHA-mediated ROS attack; however, bone
marrow stromal cells upregulated the expression of GSH in the CLL
cells and protected the cells from SAHA-induced oxidative stress
and apoptosis. The results further indicated that the GSH
precursor, NAC, protected the CLL cells against SAHA treatment.
These findings suggested that stromal cell-mediated GSH
upregulation in CLL cells is important in drug resistance to
SAHA.
PEITC is known to deplete GSH in single and
co-cultured CLL cells (8,23). The selective cytotoxic effect of
PEITC against CLL cells also makes it a potential candidate for
combination treatment with SAHA. The present study demonstrated
that SAHA induced significant ROS generation in the CLL cells,
followed by oxidative mitochondrial damage, cytochrome c
release and apoptosis, whereas stromal cells prevented SAHA-induced
ROS damage and apoptosis in the CLL cells. The combination of SAHA
and PEITC significantly increased cellular ROS accumulation and
apoptotic cell death of the CLL cells in the presence of stromal
cells. Treatment with antioxidant NAC effectively decreased ROS and
apoptosis in the CLL cells following the combination treatment of
SAHA and PEITC, indicating that the combined effect is mediated by
ROS accumulation. Notably, although the combination of SAHA and
PEITC destroyed CLL cells cultured with stromal cells, the stromal
layer was intact following treatment, indicating that the
combination had therapeutic selectivity. In addition, since CLL
cells, which no longer express p53 are often resistant to
chemotherapy and have poor clinical outcomes, the present study
also examined the combination treatment strategy in CLL cells from
p53-negative patients, and demonstrated that the p53-null CLL cells
were resistant to SAHA treatment, even without stromal cells,
consistent with the effects of p53 on drug sensitivity. However,
these cells remained sensitive to PEITC, and the combination of 2
µM SAHA and 10 µM PEITC resulted in increased cell
death in the p53-negative CLL cells co-cultured with stromal cells.
The novel treatment combination strategy, which circumvents stromal
protection and eliminates CLL cells including p53-negative CLL
cells, has significant clinical effects.
A previous report demonstrated that SAHA induces the
early increase of ROS through nicotinamide adenine dinucleotide
phosphate oxidase in leukemia cells, which results in the
translocation of Nrf2 between the cytosol and nucleus, leading to
the upregulation of glutathione-associated enzymes as a cellular
protective mechanism (12). The
Nrf2 transcription factor controls the expression of GSH-generating
enzymes, including GCLC, a rate-limiting enzyme in GSH synthesis
(28). Nrf2 localizes to the
cytoplasm in order to interact with Keap1, and oxidative
modification of Keap1 causes the release and translocation of Nrf2
between the cytoplasm and nucleus, where it binds to antioxidant
responsive elements (40). To
investigate whether SAHA-induced ROS stress initiates the
compensatory antioxidant system in CLL cells to maintain the redox
balance, the present study examined the expression levels and
activation of Nrf2 and GCLC in CLL cells following SAHA treatment.
Confocal microscopic analysis demonstrated the translocation of
Nrf2 between the cytoplasm and nucleus in the CLL cells following
treatment with SAHA. The expression levels of Nrf2 and GCLC were
also upregulated by SAHA incubation in the CLL cells. Notably,
although Nrf2 activation and enhanced expression of GCLC has been
reported to increase GSH levels in AML cells (12), the present study demonstrated that
they failed to promote GSH synthesis in the CLL cells. These data
indicated that the mechanism underlying the resistance to SAHA
treatment is different in AML and CLL cells, and the compensatory
antioxidant system activated by SAHA treatment fails to induce SAHA
resistance in CLL cells cultured alone.
Cysteine is the rate-limiting substrate for GSH
synthesis, and the predominant source of cysteine in cells is the
uptake of extracellular cysteine or cystine through specific
transporters (41). Our previous
study suggested that CLL cells exhibit limited ability to transport
cystine due to low expression levels of the Xc-cystine transporter,
and rely on stromal cells to import cystine and convert it to
cysteine, which is released into the microenvironment for the
uptake by CLL cells to enhance GSH synthesis (8). The present study demonstrated that
treatment with SAHA increased levels of GSH in the CLL cells only
in the presence of stromal cells. It is likely that the increased
expression of the GSH synthesis enzyme following SAHA incubation
and the supplementation of cysteine by stromal cells cooperated to
promote GSH synthesis in the CLL cells. Unlike CLL cells, the
expression of GCLC and levels of GSH in AML cells increase
following SAHA incubation without stromal cells (12), which may be due to the endogenous
expression of Xc-, and thus self-sufficiency of cystine in AML
cells. In CLL and AML cells, elevated GSH protects leukemia cells
from SAHA-induced cytotoxicity, and disabling this protective
mechanism using the GSH depleting agent, PEITC, significantly
sensitizes CLL cells to SAHA treatment in the stromal
environment.
The antiapoptotic protein, Mcl1, is critical in
prolonging CLL cell survival, particularly in a tumorstroma context
(42). Previous studies have
reported that a decrease in the expression of Mcl1 is essential for
the induction of apoptosis by diverse stimuli (43,44).
However, instead of decreased expression of Mcl1, the present study
observed an increase in Mcl1 following SAHA treatment in the CLL
cells, which was concordant with a previous report that
demonstrated that HDACIs induce apoptosis primarily via the
Bcl-2-antagonist killer/Mcl1/Moxa and Bcl-2 interacting mediator of
cell death signaling pathways, without decreasing the expression of
Mcl1 (45). Furthermore, the
present study demonstrated that bone marrow stromal cells or the
GSH precursor, NAC, further increased the expression of Mcl1 in the
CLL cells treated with SAHA. A previous study reported that protein
glutathionylation regulates the functions of multiple proteins
(46), and glutathionylation of
Mcl1 regulates its stability (23). Since bone marrow stromal cells
increased the levels of GSH in the CLL cells, the upregulated
protein expression of Mcl1 in the CLL cells following SAHA
treatment may be further glutathionylated and stabilized in a
stromal environment.
Mcl1 is functionally important in leukemia cells, as
marked potentiation of HDACI-induced apoptosis has been reported
following the inhibition of Mcl1 by either cyclin-dependent kinase
inhibitors or Mcl1 small interfering RNA in leukemia cells
(45). Therefore, HDACI-mediated
Mcl1 upregulation may have an important antiapoptotic role in
limiting the efficacy of HDACI-induced apoptosis. In the present
study, the results demonstrated that the GSH depleting agent,
PEITC, suppressed the expression of Mcl1 through
deglutathionylation, and circumvented drug resistance to SAHA
treatment in the CLL cells co-cultured with stromal cells. A
previous report demonstrated that the loss of p53 leads to the
upregulation of Mcl1 through the downregulation of microRNA
(miR)-15a and miR-16-1, and thereby contributes to drug resistance
in p53-null CLL cells (47). In
the present study, Mcl1 in the p53-null CLL cells was further
increased by SAHA treatment in the presence of stromal cells, and
induced marked drug resistance. Notably, compared with the
p53-positive cells, the p53-negative CLL cells co-cultured with
stromal cells were highly resistant to SAHA treatment. However,
p53-positive and negative CLL cells exhibited high sensitivity to
the combination of SAHA and PEITC treatment in the presence of
stromal cells, which was partially due to the PEITC-mediated
suppression of Mcl1 in the CLL cells.
In conclusion, the results of the present study
demonstrated that bone marrow stromal cells upregulated the levels
of GSH in CLL cells, and protected CLL cells against SAHA-induced
oxidative stress and apoptosis. Notably, the present study
demonstrated that the combination of a HDACI with the GSH depletion
agent, PEITC, enabled the induction of high levels of CLL
apoptosis, and overcame the protection of CLL cells by bone marrow
stromal cells. The mechanism underlying the combination
cytotoxicity involved disruption of the stroma-leukemia interaction
through the GSH-mediated cyto-protective signaling pathway by
PEITC, which significantly enhanced the anti-leukemic activity of
HDACI. These results suggested that SAHA in combination with PEITC
represents a promising treatment strategy to overcome
microenvironment-mediated drug resistance and improve the treatment
outcome in patients with CLL.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant no. 81201714), the
Science Foundation of the Jiangxi Science and Technology Department
(grant no. 20142BAB215043) and the Science Foundation of the
Jiangxi Educational Committee (grant no. GJJ14173).
References
|
1
|
Keating MJ, Chiorazzi N, Messmer B, Damle
RN, Allen SL;, Rai KR, Ferrarini M and Kipps TJ: Biology and
treatment of chronic lymphocytic leukemia. Hematology Am Soc
Hematol Educ Program. 2003:153–175. 2003. View Article : Google Scholar
|
|
2
|
Tam CS and Keating MJ: Chemoimmunotherapy
of chronic lymphocytic leukemia. Best Pract Res Clin Haematol.
20:479–498. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chiorazzi N, Rai KR and Ferrarini M:
Chronic lymphocytic leukemia. N Engl J Med. 352:804–815. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Munk Pedersen I and Reed J:
Microenvironmental interactions and survival of CLL B-cells. Leuk
Lymphoma. 45:2365–2372. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Silber R, Farber CM, Papadopoulos E,
Nevrla D, Liebes L, Bruck M, Brown R and Canellakis ZN: Glutathione
depletion in chronic lymphocytic leukemia B lymphocytes. Blood.
80:2038–2043. 1992.PubMed/NCBI
|
|
6
|
Collins RJ, Verschuer LA, Harmon BV,
Prentice RL, Pope JH and Kerr JF: Spontaneous programmed death
(apoptosis) of B-chronic lymphocytic leukaemia cells following
their culture in vitro. Br J Haematol. 71:343–350. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhou Y, Hileman EO, Plunkett W, Keating MJ
and Huang P: Free radical stress in chronic lymphocytic leukemia
cells and its role in cellular sensitivity to ROS-generating
anticancer agents. Blood. 101:4098–4104. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang W, Trachootham D, Liu J, Chen G,
Pelicano H, Garcia-Prieto C, Lu W, Burger JA, Croce CM, Plunkett W,
Keating MJ and Huang P: Stromal control of cystine metabolism
promotes cancer cell survival in chronic lymphocytic leukaemia. Nat
Cell Biol. 14:276–286. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kelland L: The resurgence of
platinum-based cancer chemotherapy. Nat Rev Cancer. 7:573–584.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xu WS, Parmigiani RB and Marks PA: Histone
deacetylase inhibitors: Molecular mechanisms of action. Oncogene.
26:5541–5552. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rosato RR, Almenara JA and Grant S: The
histone deacetylase inhibitor MS-275 promotes differentiation or
apoptosis in human leukemia cells through a process regulated by
generation of reactive oxygen species and induction of p21CIP1/WAF1
1. Cancer Res. 63:3637–3645. 2003.PubMed/NCBI
|
|
12
|
Hu Y, Lu W, Chen G, Zhang H, Jia Y, Wei Y,
Yang H, Zhang W, Fiskus W, Bhalla K, et al: Overcoming resistance
to histone deacetylase inhibitors in human leukemia with the redox
modulating compound β-phenylethyl isothiocyanate. Blood.
116:2732–2741. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
O'Connor OA, Heaney ML, Schwartz L,
Richardson S, Willim R, MacGregor-Cortelli B, Curly T, Moskowitz C,
Portlock C, Horwitz S, et al: Clinical experience with intravenous
and oral formulations of the novel histone deacetylase inhibitor
suberoylanilide hydroxamic acid in patients with advanced
hematologic malignancies. J Clin Oncol. 24:166–173. 2006.
View Article : Google Scholar
|
|
14
|
Stamatopoulos B, Meuleman N, De Bruyn C,
Delforge A, Bron D and Lagneaux L: The histone deacetylase
inhibitor suberoylanilide hydroxamic acid induces apoptosis,
downregulates the CXCR4 chemokine receptor and impairs migration of
chronic lymphocytic leukemia cells. Haematologica. 95:1136–1143.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pérez-Perarnau A, Coll-Mulet L,
Rubio-Patiño C, Iglesias-Serret D, Cosialls AM, González-Gironès
DM, de Frias M, de Sevilla AF, de la Banda E, Pons G and Gil J:
Analysis of apoptosis regulatory genes altered by histone
deacetylase inhibitors in chronic lymphocytic leukemia cells.
Epigenetics. 6:1228–1235. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Abou-Nassar K and Brown JR: Novel agents
for the treatment of chronic lymphocytic leukemia. Clin Adv Hematol
Oncol. 8:886–895. PubMed/NCBI
|
|
17
|
Blum KA, Advani A, Fernandez L, Van Der
Jagt R, Brandwein J, Kambhampati S, Kassis J, Davis M, Bonfils C,
Dubay M, et al: Phase II study of the histone deacetylase inhibitor
MGCD0103 in patients with previously treated chronic lymphocytic
leukaemia. Br J Haematol. 147:507–514. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Byrd JC, Marcucci G, Parthun MR, Xiao JJ,
Klisovic RB, Moran M, Lin TS, Liu S, Sklenar AR, Davis ME, et al: A
phase 1 and pharmacodynamic study of depsipeptide (FK228) in
chronic lymphocytic leukemia and acute myeloid leukemia. Blood.
105:959–967. 2005. View Article : Google Scholar
|
|
19
|
Cheson BD, Bennett JM, Grever M, Kay N,
Keating MJ, O'Brien S and Rai KR: National cancer
institute-sponsored working group guidelines for chronic
lymphocytic leukemia: revised guidelines for diagnosis and
treatment. Blood. 87:4990–4997. 1996.PubMed/NCBI
|
|
20
|
Huang P, Sandoval A, Van Den Neste E,
Keating MJ and Plunkett W: Inhibition of RNA transcription: A
biochemical mechanism of action against chronic lymphocytic
leukemia cells by fludarabine. Leukemia. 14:1405–1413. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Iverson SL and Orrenius S: The
cardiolipincytochrome c interaction and the mitochondrial
regulation of apoptosis. Arch Biochem Biophys. 423:37–46. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zenkov NK, Menshchikova EB and Tkachev VO:
Keap1/Nrf2/ARE redox-sensitive signaling system as a
pharmacological target. Biochemistry (Mosc). 78:19–36. 2013.
View Article : Google Scholar
|
|
23
|
Trachootham D, Zhang H, Zhang W, Feng L,
Du M, Zhou Y, Chen Z, Pelicano H, Plunkett W, Wierda WG, et al:
Effective elimination of fludarabine-resistant CLL cells by PEITC
through a redox-mediated mechanism. Blood. 112:1912–1922. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pelicano H, Xu RH, Du M, Feng L, Sasaki R,
Carew JS, Hu Y, Ramdas L, Hu L, Keating MJ, et al: Mitochondrial
respiration defects in cancer cells cause activation of Akt
survival pathway through a redox-mediated mechanism. J Cell Biol.
175:913–923. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ungerstedt JS, Sowa Y, Xu WS, Shao Y,
Dokmanovic M, Perez G, Ngo L, Holmgren A, Jiang X and Marks PA:
Role of thioredoxin in the response of normal and transformed cells
to histone deacetylase inhibitors. Proc Natl Acad Sci USA.
102:673–678. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Itoh K, Wakabayashi N, Katoh Y, Ishii T,
Igarashi K, Engel JD and Yamamoto M: Keap1 represses nuclear
activation of antioxidant responsive elements by Nrf2 through
binding to the amino-terminal Neh2 domain. Genes Dev. 13:76–86.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dinkova-Kostova AT, Holtzclaw WD, Cole RN,
Itoh K, Wakabayashi N, Katoh Y, Yamamoto M and Talalay P: Direct
evidence that sulfhydryl groups of Keap1 are the sensors regulating
induction of phase 2 enzymes that protect against carcinogens and
oxidants. Proc Natl Acad Sci USA. 99:11908–11913. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lu SC: Regulation of glutathione
synthesis. Curr Top Cell Regul. 36:95–116. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Craig RW: MCL1 provides a window on the
role of the BCL2 family in cell proliferation, differentiation and
tumorigenesis. Leukemia. 16:444–454. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Balakrishnan K, Burger JA, Wierda WG and
Gandhi V: AT-101 induces apoptosis in CLL B cells and overcomes
stromal cell-mediated Mcl-1 induction and drug resistance. Blood.
113:149–153. 2009. View Article : Google Scholar :
|
|
31
|
Zenz T, Häbe S, Denzel T, Mohr J, Winkler
D, Bühler A, Sarno A, Groner S, Mertens D, Busch R, et al: Detailed
analysis of p53 pathway defects in fludarabine-refractory chronic
lymphocytic leukemia (CLL): Dissecting the contribution of 17p
deletion, TP53 mutation, p53-p21 dysfunction and miR34a in a
prospective clinical trial. Blood. 114:2589–2597. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Caligaris-Cappio F: Biology of chronic
lymphocytic leukemia. Rev Clin Exp Hematol. 4:5–21. 2000.
View Article : Google Scholar
|
|
33
|
Shao Y, Gao Z, Marks PA and Jiang X:
Apoptotic and autophagic cell death induced by histone deacetylase
inhibitors. Proc Natl Acad Sci USA. 101:18030–18035. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Okabe S, Tauchi T, Kimura S, Maekawa T,
Kitahara T, Tanaka Y and Ohyashiki K: Combining the ABL1 kinase
inhibitor ponatinib and the histone deacetylase inhibitor
vorinostat: A potential treatment for BCR-ABL-positive leukemia.
PLoS One. 9:e890802014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Thurn KT, Thomas S, Moore A and Munster
PN: Rational therapeutic combinations with histone deacetylase
inhibitors for the treatment of cancer. Future Oncol. 7:263–283.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nimmanapalli R, Fuino L, Stobaugh C,
Richon V and Bhalla K: Cotreatment with the histone deacetylase
inhibitor suberoylanilide hydroxamic acid (SAHA) enhances
imatinib-induced apoptosis of Bcr-Abl-positive human acute leukemia
cells. Blood. 101:3236–3239. 2003. View Article : Google Scholar
|
|
37
|
He LZ, Tolentino T, Grayson P, Zhong S,
Warrell RP Jr, Rifkind RA, Marks PA, Richon VM and Pandolfi PP:
Histone deacetylase inhibitors induce remission in transgenic
models of therapy-resistant acute promyelocytic leukemia. J Clin
Invest. 108:1321–1330. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
VanderMolen KM, McCulloch W, Pearce CJ and
Oberlies NH: Romidepsin (Istodax, NSC 630176, FR901228, FK228,
depsipeptide): A natural product recently approved for cutaneous
T-cell lymphoma. J Antibiot (Tokyo). 64:525–531. 2011. View Article : Google Scholar
|
|
39
|
Garcia-Manero G, Yang H, Bueso-Ramos C,
Ferrajoli A, Cortes J, Wierda WG, Faderl S, Koller C, Morris G,
Rosner G, et al: Phase 1 study of the histone deacetylase inhibitor
vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients
with advanced leukemias and myelodysplastic syndromes. Blood.
111:1060–1066. 2008. View Article : Google Scholar
|
|
40
|
Kansanen E, Kuosmanen SM, Leinonen H and
Levonen AL: The Keap1-Nrf2 pathway: Mechanisms of activation and
dysregulation in cancer. Redox Biol. 1:45–49. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bannai S: Transport of cystine and
cysteine in mammalian cells. Biochim Biophys Acta. 779:289–306.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kitada S, Andersen J, Akar S, Zapata JM,
Takayama S, Krajewski S, Wang HG, Zhang X, Bullrich F, Croce CM, et
al: Expression of apoptosis-regulating proteins in chronic
lymphocytic leukemia: Correlations with in vitro and in vivo
chemoresponses. Blood. 91:3379–3389. 1998.PubMed/NCBI
|
|
43
|
Nijhawan D, Fang M, Traer E, Zhong Q, Gao
W, Du F and Wang X: Elimination of Mcl-1 is required for the
initiation of apoptosis following ultraviolet irradiation. Genes
Dev. 17:1475–1486. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Adams KW and Cooper GM: Rapid turnover of
mcl-1 couples translation to cell survival and apoptosis. J Biol
Chem. 282:6192–6200. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Inoue S, Walewska R, Dyer MJ and Cohen GM:
Downregulation of Mcl-1 potentiates HDACi-mediated apoptosis in
leukemic cells. Leukemia. 22:819–825. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ghezzi P: Regulation of protein function
by glutathionylation. Free Radic Res. 39:573–580. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
An X, Schulz VP, Li J, Wu K, Liu J, Xue F,
Hu J, Mohandas N and Gallagher PG: Global transcriptome analyses of
human and murine terminal erythroid differentiation. Blood.
123:3466–3477. 2014. View Article : Google Scholar : PubMed/NCBI
|