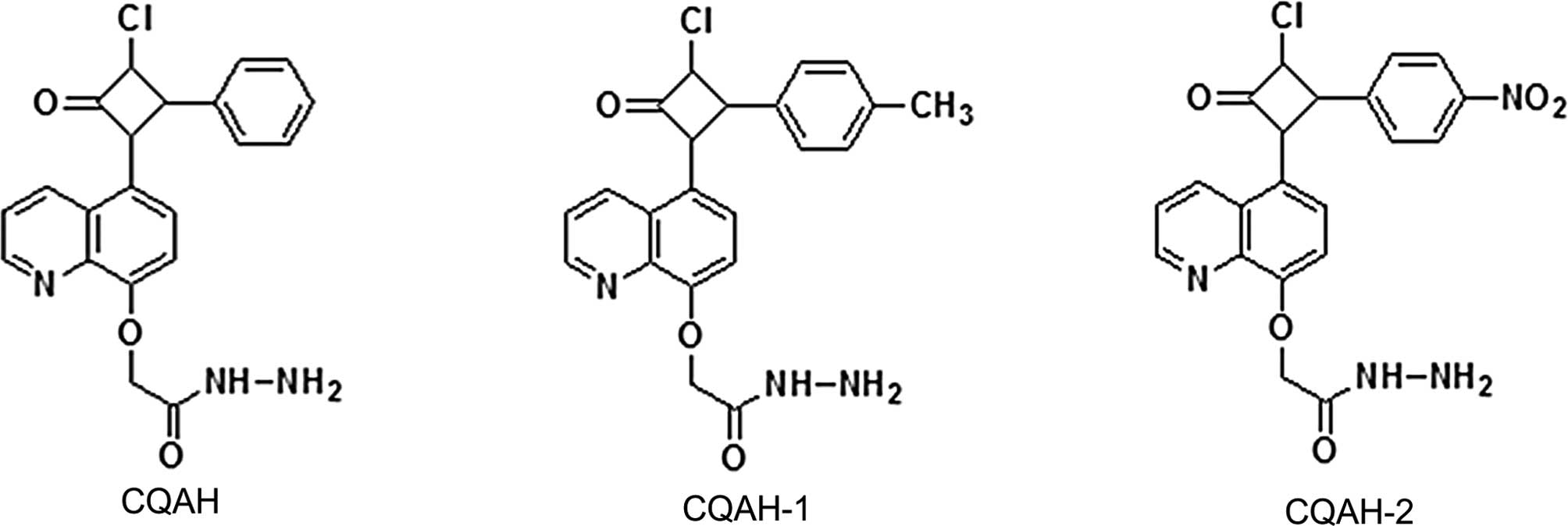
Introduction
Cancer is one of the greatest challenges in the
clinical field worldwide and its occurrence is increasing in
developed as well as in developing countries. In the United States,
colorectal cancer is considered to be the second-largest cause of
cancer-associated mortality (1,2). In
spite of the fact that the genetics of colon cancer have been
studied in depth (3), current
therapies are not able to effectively treat colorectal cancer
(4,5).
Although colorectal cancer can be cured at early
stages, patients frequently present with metastases at the
time-point of the occurrence of symptoms and diagnosis, leading to
a high mortality rate (6). Hence,
research efforts focus on developing novel and more potent
preventive and screening methods for colon cancer (7). Numerous studies have shown that the
mortality rate arising from colorectal cancer decreased to 40–50%
in patients with colorectal cancer taking non-steroidal
anti-inflammatory drugs and aspirin (8–13),
clearly demonstrating the chemopreventive effects of these drugs.
Other studies reported that sunlindac is a potent drug causing a
regression of the adenoma count and size in patients with
adenomatous polyposis (14–17).
The primary method of colon cancer treatment is surgery.
5-fluoruracil (5-FU), camptothecin-11 (CPT-11; irinotecan) and
oxaliplatin are the most common chemotherapeutic drugs used to
destroy cancer cells after tumor resection (18,19).
Combinatorial treatment of drug administration and surgery has
enhanced therapeutic efficacy as compared with that of monotherapy
(20,21). Besides from irradiation and
surgical treatments, chemotherapy remains one of the primary
choices for cancer treatment (22).
Quinoline moieties and their oxo-derivatives have
gained substantial interest due to their occurrence in a wide range
of natural products and bio-active compounds (23). 3-substituted quinoline-2-one is a
key moiety present in a variety of compounds with anti-cancer
properties, and quin-oline-2-one-based compounds were found to be
effective and promising lead structures for kinase inhibition
(24).
Azetidinones are part of the structural skeleton of
several antibiotics and are widely known to exhibit potent
biological activities (25).
Synthesis of azetidinone derivatives has provided variety of novel
compounds with applications including anti-bacterial,
antimicrobial, anti-convulsant, anti-inflammatory and
anti-tuberculosis properties (26–30).
They can also act as enzyme inhibitors and exert effects on the
central nervous system (31).
Another class of heterocyclic compound, 8-hydroxy quinolines, has
been demonstrated to have valuable biological activities, with
derivatives being used as effective human immunodeficiency virus-1
intergrase inhibitors (32,33),
as antimicrobial compounds or as herbicides (34–36).
Quinoline-based synthetic azetidinone derivatives have also shown
potential anti-microbial properties (37,38).
Based on the abovementioned findings, the present study
investigated the anti-cancer effects of compounds containing
azetidine and the quinoline-2-one skeleton combined in one
molecule. Madhu et al (39)
have previously synthesized
[5-(3-chloro-oxo-4-phenyl-cyclobutyl)-quinoli-8-yl-oxy] acetic acid
hydrazide (CQAH), which showed efficacy against a range of bacteria
and fungi. The present study explored the effects of CQAH as well
as two if its derivatives, CQAH-1 and -2, bearing a methyl- or
nitro-substitution, respectively, at position-4 of the azetidine
phenyl ring (Fig. 1), on
colorectal cancer in order to assess their potential for use as
novel chemotherapeutic drugs.
Materials and methods
Compounds and reagents
CQAH and its derivatives were prepared according to
the procedure of a previous study (39). The identity of the compounds was
confirmed by infrared (IR) spectroscopy and nuclear magnetic
resonance (NMR). The spectroscopical data, with assignments as
singulet (s), multiplet (m) and duplet (d) were as follows:
2-((5-(3-chloro-2-oxo-4-phenylazetidin-1-yl)
quinolin-8-yl)oxy)acetohydrazide (CQAH). IR (KBr) in
cm−1: 674 (–Cl), 1,625 (–C=N), 1,685 (–C=O), 3,210
(–NH), 3,410 (–NH2), 3,486. 1HNMR [300 MHz,
(CD3)2SO, tetramethylsilane (TMS)]:
δ=2.09 (s, 2H, –NH2), 4.7 (s, 2H,
–O–CH2), 5.12 (d, 1H, –CH of azetidin attached to
phenyl), 5.41 (d, 1H, –CH of azetidin attached to –Cl), 6.55 (d,
1H, –CH), 6.78 (d, 1H, –CH), 7.1–7.2 (m, 5H of
C6H5), 7.8–8.8 (m, 3H of quinoline ring),
10.08 (s, 1H, –NH). 13C NMR (75 MHz, CDCl3,
TMS) δ=60 (C–Cl), 63 (N–CH–Ar), 68 (O–CH2), 108,
115, 119, 121, 127 (Ar–C), 128, 129, 138, 134, 141, 147, 150, 161
(N–C=O), 167 (–CO–N).
2-(5-(3-chloro-2-oxo-4-p-tolylazetidin-1-yl)
quinolin-8-yloxy)acetohydrazide (CQAH–1). IR (KBr) in
cm−1: 678 (–Cl), 1,620 (–C=N), 1,684 (–C=O), 3,208
(–NH), 3,410 (–NH2), 3,494. 1HNMR [300 MHZ,
(CD3)2SO, TMS]: δ=2.10 (s, 2H,
–NH2), 2.30 (s, 3H, Ar–CH3), 4.73 (s, 2H,
–O–CH2), 5.14 (d, 1H azetidin–CH attached to phenyl),
5.45 (d, 1H, azetidin–CH attached to Cl), 6.3 (d, 1H, –CH), 6.4 (d,
1H, –CH),7.0 (m, 4H of C6H4), 8–8.7 (m, 3H of
quinoline ring), 10.08 (s, 1H, –NH). 13C NMR (75 MHz,
CDCl3, TMS) δ=25 (Ar–CH3), 62 (C–Cl),
65 (N–CH–Ar), 67 (O–CH2), 114, 117, 123, 127, 128, 130,
134 (Ar–C), 135, 137, 139, 147, 150, 162 (N–C=O), 167, 168
(–CO–N).
2-((5-(3-chloro-2-(4-nitrophenyl)-4-oxoazetidin-1-yl)
quinolin-8-yl)oxy) acetohydrazide (CQAH–2). IR (KBr) in
cm−1: 675 (–Cl), 1,614 (–C=N), 1,680 (–C=O), 3,208
(–NH), 3,412 (–NH2), 3,494. 1H NMR [300 MHZ,
(CD3)2SO, TMS]: δ=2.10 (s, 2H,
–NH2), 4.80 (s, 2H, –O–CH2), 5.13 (d, 1H, –CH
of azetidin attached to phenyl ring), 5.46 (d, 1H, –CH of azetidin
attached to –Cl), 6.54 (d, 1H, –CH), 6.74 (d, 1H, –CH), 7.0–8.0 (m,
4H of C6H4), 7.3–8.8 (m, 3H of quinoline
ring), 10.10 (s, 1H, –NH). 13C NMR (75 MHz,
CDCl3, TMS) δ=62 (C–Cl), 65 (N–CH–Ar), 67
(O–CH2), 106, 117, 120, 122, 127, 129, 134, 138, 147
(Ar–C), 151, 153, 162 (N–C=O), 167 (–CO–N).
All chemicals used in the present study were
purchased from Sigma-Aldrich (St. Louis, MO, USA). Diphenylene
iodonium (DPI), MTT and N-acetyl-l-cysteine (NAC) were obtained
from Sigma-Aldrich. Antibodies against c-Jun N-terminal kinase
(JNK; cat no. 8528; 1:5,000; rabbit polyclonal IgG, 1 h at 25°C)
and phosphorylated (p)-JNK (cat no. 4668; 1:2,000; rabbit
polyclonal IgG, 1 h at 25°C) were obtained from Cell Signaling
Technology, Inc. (Beverly, MA, USA). Antibodies against B-cell
lymphoma (Bcl) extra-large protein (Bcl-XL; cat no. GTX100632;
1:3,000; rabbit polyclonal IgG, 1 h at 25°C), Bcl-2 homologous
antagonist killer (Bak; cat. no. GTX100063; 1:3,000; rabbit
polyclonal IgG, 1 h at 25°C), poly(adenosine diphosphate ribose)
polymerase (PARP; cat no. GTX100573; 1:3,000; rabbit polyclonal
IgG, 1 h at 25°C), caspase-3 (cat. no. GTX110543; 1:3,000; rabbit
polyclonal IgG, 1 h at 25°C), myeloid cell leukemia 1 (Mcl-1; cat.
no. GTX102026; 1:1,000; rabbit polyclonal IgG, 1 h at 25°C) and
α-tubulin (cat. no. GTX112141; 1:10,000; rabbit polyclonal IgG, 1 h
at 25°C) were purchased from Gene Tex (San Antonio, TX, USA).
Mitogen-activated protein kinase (MAPK) inhibitors, including
extracellular signal-regulated kinase (ERK) inhibitor PD98059, p38
inhibitor SB203580 and JNK inhibitor SP600125 were obtained from
Merck Millipore (Billerica, MA, USA).
Cell culture and MTT assay
The HCT116 and LoVo human colorectal cancer cell
lines were purchased from the American type culture collection
(Manassas, VA, USA). These cells were maintained in Dulbecco's
modified Eagle's medium (Sigma-Aldrich) supplemented with 5%
heat-activated fetal bovine serum (FBS), penicillin-streptomycin
(100 U) and sodium pyruvate (1 mM) in a humidified incubator with
5% CO2 at 37°C.
For the MTT assay, HCT116 and LoVo cells seeded in
24-well plates at a density of 1×105 per well in growth
medium (Sigma-Aldrich). Subsequent to the cells being re-fed with
growth medium, they were incubated with various concentrations of
CQAH (0–20 μM) for 24 or 48 h. The growth medium was fully
removed and the cells were incubated with MTT solution (50
μg/ml) for 2 h. The obtained formazan crystals were
dissolved in isopropanol and the absorbance was recorded at a
wavelength of 560 nm using an ELISA reader (SpectraMax 190;
Molecular Devices Inc., Sunnyvale, CA, USA). The cell viability was
determined as a percentage of the control.
In order to determine the role of the MAPK pathway
in drug-induced apoptosis, cells were pretreated with inhibitors of
ERK (PD98059; 10 and 20 μM); JNK (SP600125; 10 and 20
μM) or p38 (SB203580; 10 and 20 μM). CQAH (10
μM) treatment was performed for 48 h and the viability of
the cells was measured using an MTT assay.
The cells were treated with the antioxidant agents
NAC (glutathione activator) and DPI (NAPDH inhibitor) to determine
their role in drug-induced cell death. Cells were pretreated with
inhibitors of glutathione (NAC; 5 and 10 μM) or NADPH (DPI;
5 and 10 μM). CQAH (10 μM) was added and cells were
incubated for 48 h. The viability of the cells was assessed using
an MTT assay.
Assessment of apoptosis
Cells at a density of 1×105 per well were
seeded into wells containing glass slips and treated with CQAH (10
μM) for 24 h. The slips were fixed with methanol for 10 min
and treated with DAPI for 30 min. Slides were mounted with mounting
medium (Santa Cruz Biotechnology, Inc., Dallas, TX, USA). The
percentage of apoptotic nuclei was determined by counting condensed
and bright nuclei compared with the total number of cells.
In another experiment, the cells were pretreated
with inhibitors of caspase-3 (Z-DEVD-FMK; 50 and 100 μM) or
caspase-9 (Z-LEHD-FMK; 50 and 100 μM) to inhibit the action
of apoptosis-associated enzymes. CQAH (10 μM) was then added
for 48 h. The cell viability was measured using an MTT assay.
Annexin V-propidium iodide (PI)
staining
An Annexin-V-fluorescein isothiocyanate
(FITC)/double staining kit (BD Biosciences, Franklin Lakes, NJ,
USA) was used to quantify the apoptosis of CQAH-treated HCT116 and
LoVo cells. In brief, HCT116 and LoVo cells at a density of
1×105 per well were seeded in 24-well plates and then
treated with CQAH at various concentrations (0–20 μM) for 24
h. The cells were then trypsinized, treated with binding buffer and
Annexin-FITC and PI were added, followed by incubation for 15 min
in the dark according to the manufacturer's instructions of the
apoptosis kit. The cells were analyzed using a FACSCalibur flow
cytometer (BD Biosciences).
Western blot analysis
For western blot analysis, HCT116 and LoVo cells
were washed two times using ice-cold phosphate-buffered saline
(PBS) and then extracted in radioimmunoprecipitation assay buffer
containing Tris-HCl (50 mM, pH 7.4), 1% nonidet P-40, NaCl (150
mM), ethylene glycol tetraacetic acid (1 mM), sodium deoxycholate
(0.025%), sodium orthovanadate (1 mM), phenylmethyl
sulfonylfluoride (1 mM), and NaF (1 mM). Cell extracts were
examined with the Bio-Rad protein assay kit (Bio-Rad Laboratories,
Inc., Hercules, CA, USA) in order to quantify the protein
concentration using bovine serum albumin (BSA) as a standard. The
ELISA reader was used to measure the absorbance at a wavelength of
595 nm. Equal quantities of protein (50 μg) separated by 8%
SDS-PAGE and then transferred onto immobilon polyvinylidene
difluoride membranes (Merck-Millipore). 1% BSA was used to block
the membranes at room temperature, and membranes were subsequently
incubated with specific primary antibodies overnight. Membranes
were washed three times in PBS containing Tween 20, followed by
incubation with horseradish peroxidase-conjugated secondary
antibody for 1 h. The protein expression was visualized using an
enhanced chemiluminescence assay kit (cat. no. WBKLS0500;
Merck-Millipore) by ImageQuant LAS4000 (GE Healthcare Bio-Sciences,
Pittsburgh, PA, USA).
In order to determine the role of the MAPK pathway
in drug-induced apoptosis, cells were pretreated with inhibitors of
ERK (PD98059; 10 and 20 μM); JNK (SP600125; 10 and 20
μM) or p38 (SB203580; 10 and 20 μM). CQAH (10
μM) treatment was performed for 48 h. The protein expression
of MAPK-associated proteins was assessed using western blot
analysis.
In order to determine the role of antioxidants in
drug-induced cell death, cells were treated with NAC (10 μM)
and DPI (5 μM) in the presence of CQAH (10 μM) for 24
h, and the protein expression of PARP and caspase-3 was analyzed
using western blotting.
Transient transfection
HCT116 and LoVo cells were first seeded in 6-cm
dishes and transfection was performed using dominant-negative JNK
(DN-JNK) or pcDNA3 with PolyJect reagent according to the
manufacturer's instructions (SignaGen Laboratories, Gaithersburg,
MD, USA). After 24 h of transfection, cells were trypsinized,
seeded in a 24-well plate and then treated with CQAH for 48 h.
Statistical analysis
All experimental data are from three independent
experiments and values are expressed as the mean ± standard error.
Statistical analysis was conducted with GraphPad Prism, version 5.0
(GraphPad Software, Inc., La Jolla, CA, USA). Differences between
groups were assayed using Student's t-test for each paired
experiment. P<0.05 was considered to indicate a statistically
significant difference between values.
Results
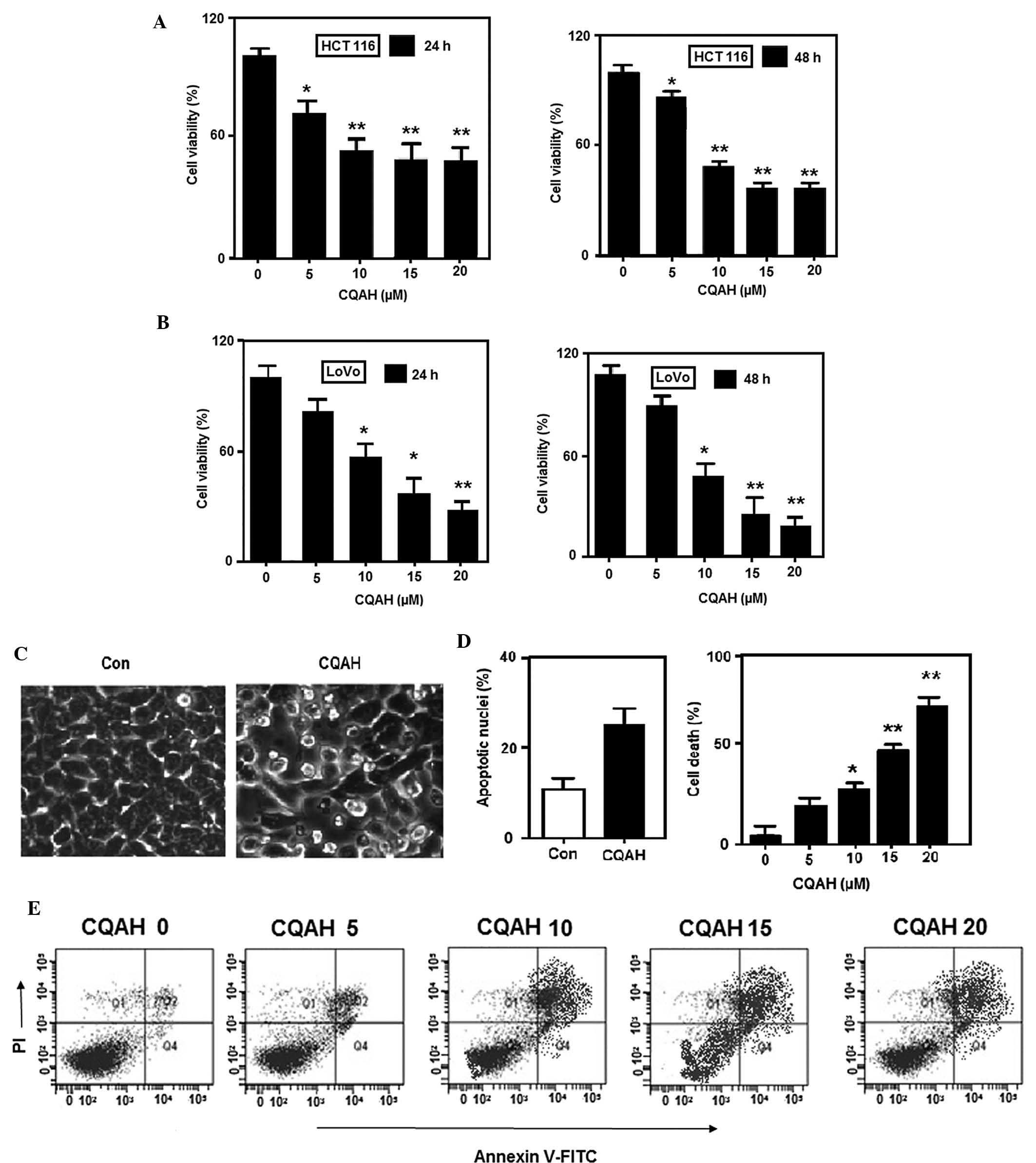
CQAH decreases the viability of HCT116
and LoVo human colorectal cancer cells by inducing apoptosis
CQAH and its analogues (Fig. 1) have been previously shown to
exhibit anti-bacterial activity against Escherichia coli,
Bacillus cereus, Staphylococcus aureus and
Pseudomonas aeruginosa (39); however, their effects on cancer
have not been elucidated, to the best of our knowledge. In order to
assess the effects of CQAH on colon cancer, the HCT116 and LoVo
cell lines were treated with CQAH for 24 or 48 h and subjected to
MTT assays. CQAH exerted considerable cytotoxic effects on the two
cell lines in a dose-dependent manner (Fig. 2A and B) with 48-h
IC50-values of ~10 μM for HCT116 as well as LoVo.
In order to assess whether CQAH can induce tumor-cell apoptosis,
HCT116 cells were treated with CQAH and morphological changes were
observed by microscopy (Leica TCS SP8 STED 3X; Leica Microsystems,
Wetzlar, Germany), indicating that cell shrinkage and the formation
of apoptotic bodies were induced by CQAH treatment. To further
verify that apoptosis was induced by CQAH, chromatin condensation
was observed by staining the cells with DAPI (Fig. 2C), revealing that CQAH treatment
produced a marked increase in chromatin condensation and
considerably increased the amount of apoptotic nuclei from 10 to
30% (Fig. 2D). Furthermore,
Annexin V-PI staining and flow cytometric analysis demonstrated an
increase in the apoptotic rate from 20 to 85% upon CQAH treatment
(Fig. 2E).
CQAH induces apoptosis-associated
signaling in colorectal cancer cells
To examine the apoptotic pathways activated by CQAH
in colorectal cancer cells, the present study assessed the effects
of CQAH on two key apoptotic proteins, caspase-3 and PARP, which
participate in a proteolytic signaling cascade that is activated
during the apoptotic process. Western blot analysis indicated that
treatment of HCT116 cells with CQAH at 0–20 μM induced
apoptosis in a concentration-dependent manner by enhancing cleaved
caspase-3 and PARP (Fig. 3A).
Time-dependent analysis revealed that pro-caspase-3 was reduced
within 16 h of incubation with CQAH, whereas increased levels of
cleaved caspase-3 and PARP where identified following CQAH
treatment for 16–32 h (Fig. 3A).
Furthermore, upon CQAH treatment, an increase in the levels of
pro-apoptotic protein Bak and a decrease of anti-apoptotic proteins
Bcl-XL and Mcl-1 were observed (Fig.
3B). In addition, pre-treatment with caspase-3-specific
inhibitor z-DEVD-FMK and caspase-9-specific inhibitor z-LEDH-FMK
significantly decreased CQAH-induced cell death (Fig. 3C).
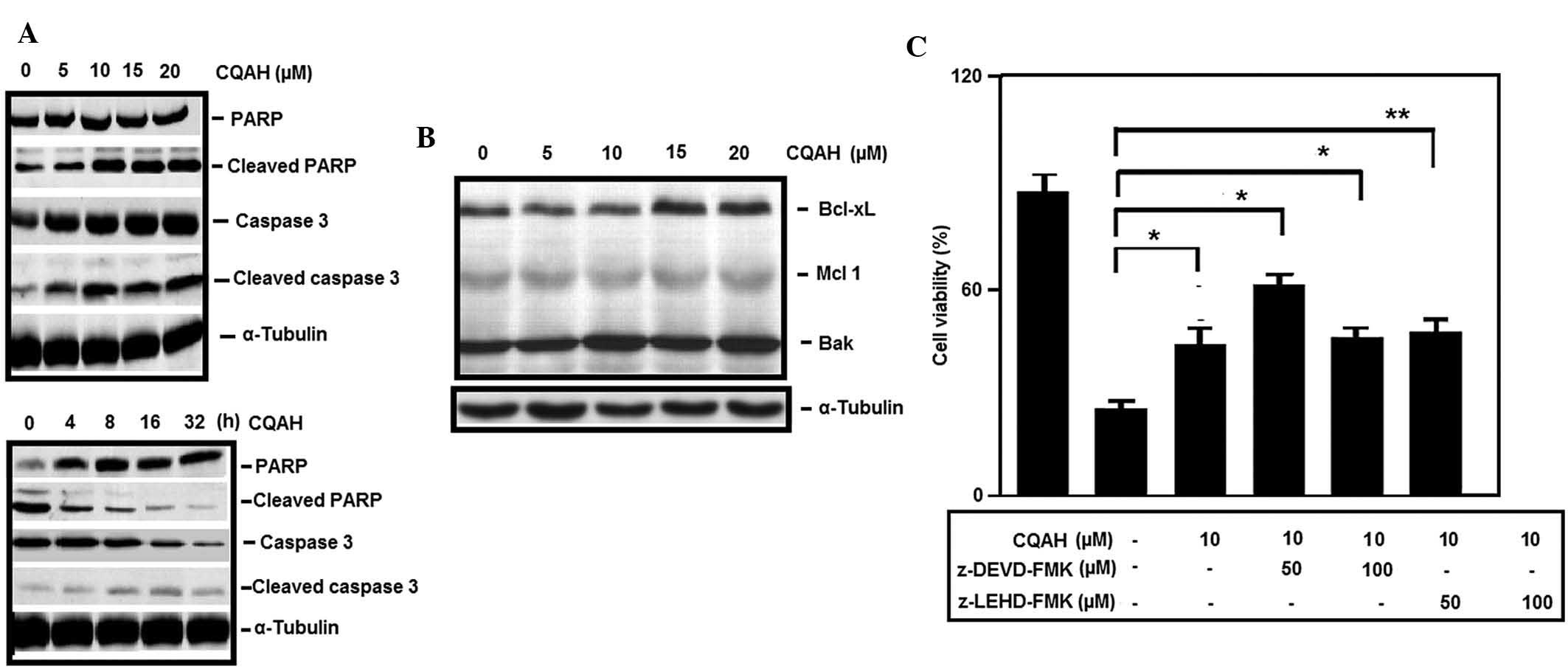 | Figure 3CQAH induces the expression of
apoptotic proteins. (A) Concentration- and time-dependent induction
of PARP and caspase-3 cleavage in the presence of CQAH. HCT116
cells were treated with 0–20 μM CQAH for 24 h or with 10
μM CQAH for 0, 4, 8, 16 and 32 h, and caspase-3 and PARP
expressions were assessed using western blot analysis. α-Tubulin
was used as the loading control. (B) HCT116 cells were treated with
CQAH (0–20 μM) for 24 h and the expression of Bcl-2 family
members Mcl-1, Bcl-XL and Bak was assessed using western blot
analysis. (C) Cells were pre-treated with inhibitors of caspase-3
(z-DEVD-FMK; 50 and 100 μM) or caspase-9 (z-LEHD-FMK; 50 and
100 μM). CQAH (10 μM) was then added for 48 h. The
cell viability was measured using an MTT assay. Data were obtained
from at least three replicates of three independent experiments,
and are expressed as the mean ± standard error.
*P<0.05; **P<0.01 as indicated. PARP,
poly(adenosine diphosphate ribose) polymerase; Bcl-2, B-cell
lymphoma 2; Bcl-XL, Bcl extra large; Bak, Bcl-2 homologous
antagonist killer; Mcl, myeloid cell leukemia. |
CQAH-induced apoptosis is mediated via
JNK activation
It is well known that MAPKs are broadly involved in
physiological regulatory processes, which are responsible for the
transduction of intracellular signaling (40). ERK, p38 and JNK have key roles in
regulating cell growth and death upon MAPK activation. The present
study investigated whether MAPK was involved in CQAH-induced
apoptosis. For this, HCT116 cells were pre-treated with the ERK,
JNK and p38 inhibitors PD98059, SP600125 and SB203580,
respectively, followed by CQAH treatment. As shown in Fig. 4A, only JNK inhibition, but not p38
or ERK inhibition, significantly reduced CQAH-induced cell death.
Furthermore, western blot analysis indicated that treatment with
JNK inhibitor SP600125 abrogated CQAH-mediated PARP and caspase-3
cleavage (Fig. 4B), while p38 and
ERK inhibitors had no marked effects. Morphological changes also
confirmed that only SP600125 efficiently prevented CQAH-induced
cell shrinkage and apoptotic body formation, as shown in Fig. 4E. Furthermore, intracellular
oxidative stress triggered the involvement of chemopreventive
product-induced cell death, which has a critical role in the
activation of MAPK and hence, the present study examined the
participation of reactive oxygen species (ROS) formation. As
indicated in Fig. 4C, the
anti-oxidant agents NAC (glutathione activator) and DPI (NAPDH
inhibitor) failed to prevent CQAH-induced cell death. Similarly,
the anti-oxidants failed to block the cleavage of PARP and
caspase-3 (Fig. 4D) or apoptotic
body formation (Fig. 4E) following
treatment with CQAH. Since JNK inhibition significantly decreased
CQAH-induced apoptosis, the present study further investigated the
involvement of JNK by assessing JNK phosphorylation following
treatment with CQAH for 4 h (Fig.
5A). Pre-treatment with SP600125 reduced CQAH-mediated
phosphorylation of JNK (Fig. 5B)
in parallel with an elevated expression of Bcl-XL protein (Fig. 5C). In addition, the cytotoxic
effects of CQAH were restored following transfection with DN-JNK
(Fig. 5D), further demonstrating
the involvement of the JNK pathway in CQAH-induced
cytotoxicity.
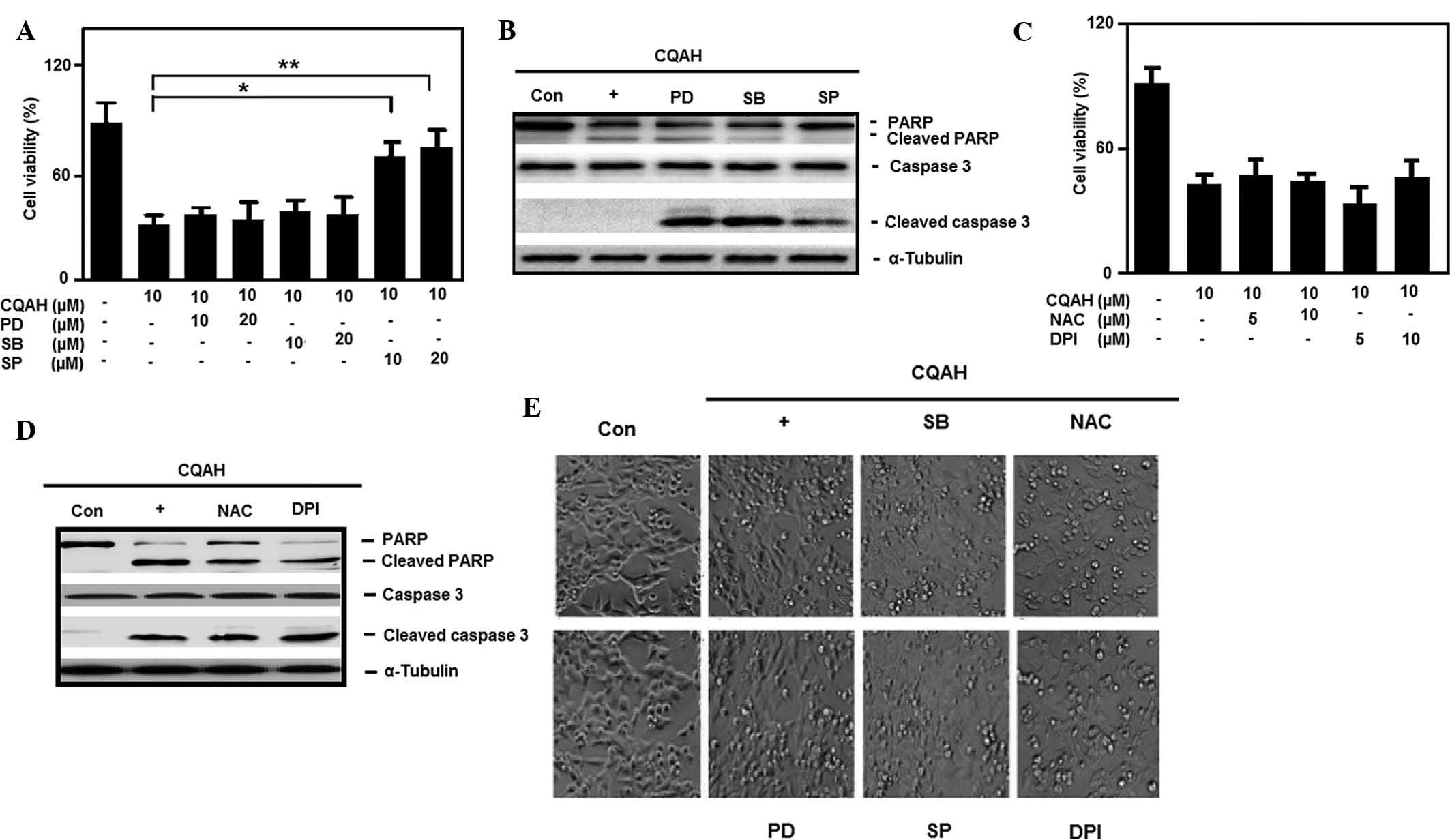 | Figure 4JNK activation-mediated CQAH
induction of apoptosis, independent of reactive oxygen species
involvement. (A) Cells were pre-treated with inhibitors of
extracellular signal-regulated kinase (PD; 10 and 20 μM),
JNK (SP; 10 and 20 μM) or p38 (SB; 10 and 20 μM).
CQAH (10 μM) treatment was performed for 48 h and the
viability of the cells was measured using an MTT assay. (B) Cells
were pre-treated with 10 μM PD, SP or SB. CQAH (10
μM) treatment was performed for 24 h, and protein expression
of PARP and caspase-3 was assessed using western blot analysis. (C)
Cells were pre-treated with inhibitors of glutathione (NAC; 5 and
10 mM) or NADPH (DPI; 5 and 10 μM), CQAH (10 μM) was
added, and cells were incubated for 48 h. The viability of the
cells was assessed using an MTT assay. (D) Cells were treated with
NAC (10 mM) and DPI (5 μM) in the presence of CQAH (10
μM) for 24 h, and protein expression of PARP and caspase-3
was analyzed using western blotting. (E) Bright-field images of
cellular morphology were captured in the presence of the indicated
inhibitors (20 μM PD, 20 μM SP, 20 μM SB, 10
mM NAC or 5 μM DPI) and CQAH (10 μM) for 48 h
(magnification, ×100). Values are expressed as the mean ± standard
error. *P<0.05; **P<0.01 as indicated.
JNK, c-Jun N-terminal kinase; PD, PD98059; SP, SP600125; SB,
SB203580; NAC, N-acetyl-l-cysteine; DPI, diphenylene
iodonium; Con, control; PARP, poly(adenosine diphosphate ribose)
polymerase. |
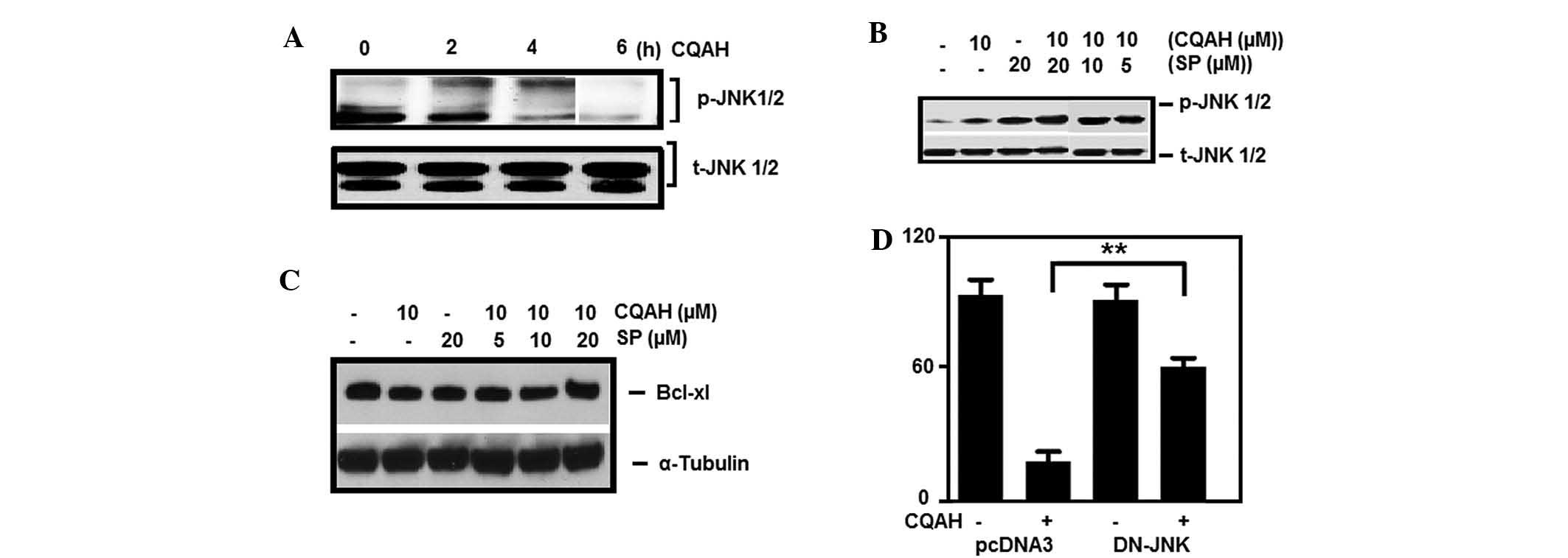 | Figure 5Activation of JNK is crucial for
CQAH-induced apoptosis. (A) HCT116 cells were treated with CQAH (10
μM) for 1–6 h, and phosphorylation of JNK was analyzed using
western blotting. Cells were pre-treated with SP600125 (5, 10 and
20 μM) and then incubated with CQAH (10 μM) for 2 h
to detect (B) p-JNK and for 24 h to detect (C) Mcl-1 and Bcl-XL.
(D) Cells were transiently transfected with DN-JNK for 18 h and
then treated with CQAH for 48 h. Cell viability was measured using
an MTT assay. Data were obtained from at least 3 replicates of 3
independent experiments, and are expressed as the mean ± standard
error. **P<0.01. p/t-JNK, phosphorylated/total c-Jun
N-terminal kinase; Bcl-2, B-cell lymphoma 2; Bcl-XL, Bcl extra
large; Bak, Bcl-2 homologous antagonist killer; Mcl, myeloid cell
leukemia; SP, SP600125; DN, dominant-negative. |
CQAH enhances the therapeutic efficacy of
5-FU and CPT-11
The present study further demonstrated the
synergistic effects of CQAH upon combined treatment with the
pyrimidine analogue anti-colon cancer drugs 5-FU and CPT-11, which
act via blocking thymidylate synthase and inhibiting topoisomerase
I (41). The results clearly
indicated that the cell viability was considerably reduced upon
combination with 5-FU or CPT11 (Fig.
6A). Accordingly, western blot analysis showed that combined
treatment of CQAH with 5-FU or CPT-11 enhanced caspase-3 and PARP
cleavage (Fig. 6B). Furthermore,
the present study examined the structure-activity association of
two derivatives of CQAH, CQAH-1 and CQAH-2. CQAH-1 carries a methyl
group at position-4 of the azetidine phenyl ring, whereas CQAH-2
carries a nitro group in the same position. Morphological
observation, MTT assay and western blot analysis indicated that
CQAH-1 treatment at a concentration of 20 μM had a higher
cytotoxic effect compared to that of CQAH, whereas CQAH-2 displayed
reduced cytotoxicity (Fig.
6C–E).
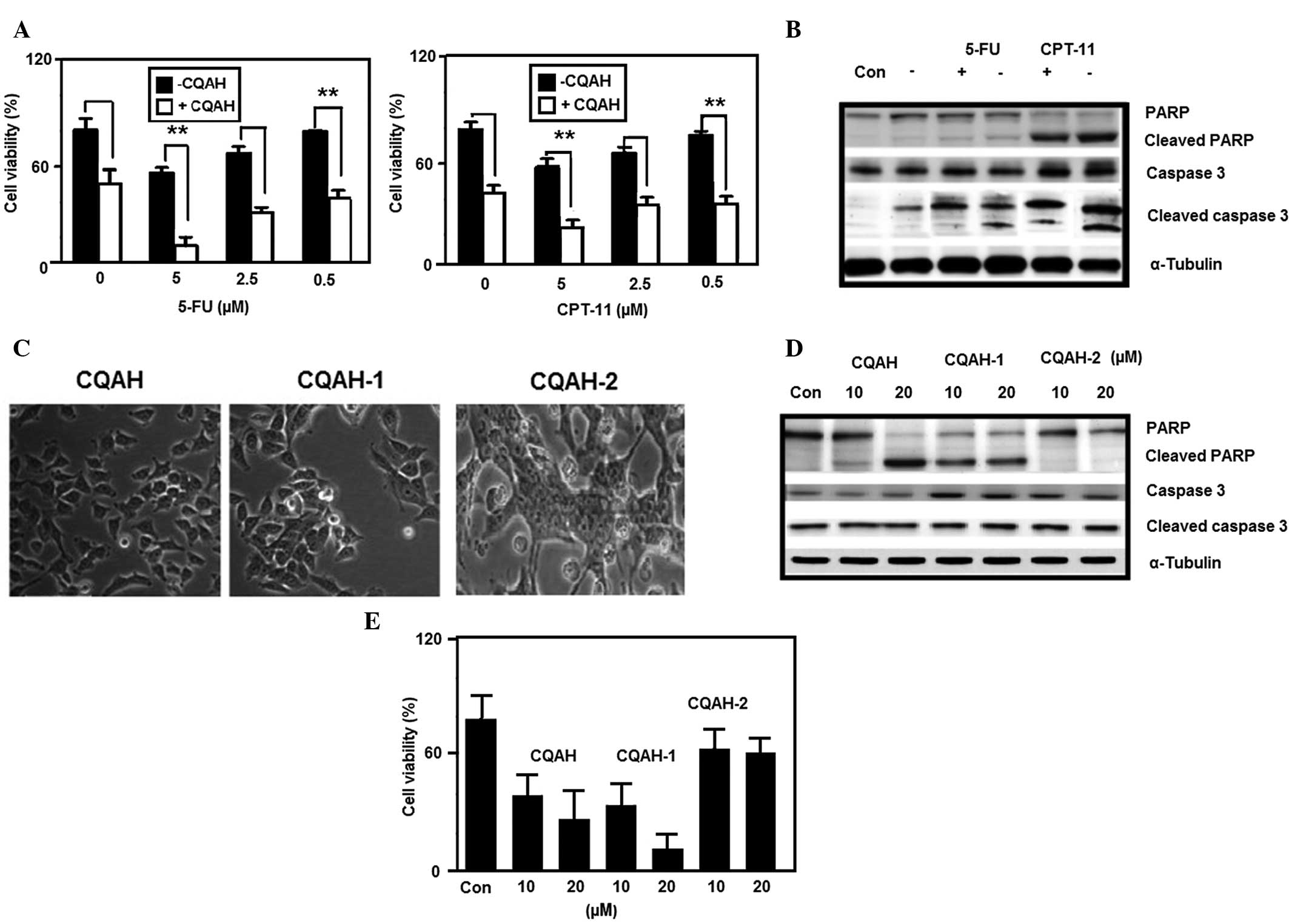 | Figure 6CQAH potentiates the therapeutic
efficacy in combination with 5-FU and CPT-11, and its analogues
CQAH-1 and CQAH-2 exert apoptotic effects on HCT116 cells. (A)
Cells were treated with CPT-11 or 5-FU (0, 5, 2.5 or 0.5 μM)
in the presence or absence of CQAH (10 μM) for 24 h, and
cell viability was assessed using an MTT assay. (B) Cells were
treated with CPT-11 or 5-FU (5 μM) in the presence or
absence of CQAH (10 μM) for 24 h, and caspase-3 and PARP
protein expressions were analyzed using western blotting. (C) Cells
were treated with CQAH, CQAH-1 or CQAH-2 (10 μM) for 48 h
and the cell morphology was observed using a light microscope
(magnification, ×100). (D) Cells were treated with CQAH, CQAH-1 or
CQAH-2 (10 or 20 μM) for 24 h and the expression of PARP and
cleaved caspase-3 was analyzed using western blotting. (E) Cell
viability was assessed using an MTT assay after 24 h of incubation.
Data were obtained from at least three replicates of three
independent experiments, and are expressed as the mean ± standard
error. **P<0.01. 5-FU, 5-fluorouracil; PARP,
poly(adenosine diphosphate ribose) polymerase; CPT-11,
camptothecin-11; Con, control. |
Discussion
The MAPK signaling pathway regulates a variety of
cellular responses with the aid of numerous intracellular and
extracellular stimuli (42). The
status of MAPK signaling controls cell fate, including
proliferation and apoptosis. The present study demonstrated that
treatment with CQAH triggered phosphorylation of JNK, whereas
inhibition of JNK blocked CQAH-mediated phosphorylation of JNK as
well as cleavage of caspase-3 and PARP; however, the mechanism of
CQAH-mediated phosphorylation of JNK remains elusive. In spite of
oxidative stress being a common trigger of cell death upstream of
MAPK, participation of ROS generation was not identified following
CQAH treatment, since pre-treatment with anti-oxidant agents did
not affect CQAH-mediated cell death. Certain quinoline derivatives
are capable of inhibiting MAPK, which activates the pro-apoptotic
JNK and p38 signaling pathways (43). In a previous study, screening for
potential kinase inhibitors identified a series of dihydropyrrolo
pyrazole quinolines as effective potential inhibitors of the
catalytic activity of MLK7 in vitro (44). A reduction in the activation of JNK
and p38 with no evident effect on the activation of ERK indicated
that this compound type may be suitable for blocking MLK7-mediated
activation of the MAPK signaling pathway in vivo, while the
underlying mechanism has remained elusive. JNK activation usually
induces p53 expression, which has a major role in cell cycle
regulation and activation of pro-apoptotic proteins; however, in
the present study, it was observed that p53 protein levels were not
altered upon CQAH treatment (data not shown), implying that
CQAH-mediated apoptosis p53-independent.
Experimental results demonstrated that microtubules
inhibition by paclitaxel and vincristine end up in elevation of
phosphorylation of JNK and eventually Bcl-2 phosphorylation which
is also unassociated with p53 expression (45). In contrast, oxidative stress
dependent apoptosis mediated through arsenite is p53 dependent and
JNK independent (46). Further,
JNK activation mediated apoptosis by direct phosphorylation of Bcl
which includes Bcl-XL, Bcl-2, Bad and Bim (47–49).
ROS are involved with the upstream signaling of all
members of the MAPK family. Han et al (50) reported that ROS-mediated JNK and
p38 activation are necessary for sanguinarine-induced HCT116 cell
death. Furthermore, Zhao et al (51) identified that JNK phosphorylation
and not p38 was involved downstream of ROS signaling as part of the
mechanism of olaquindox-induced apoptosis of HepG2 cells. In the
present study, ROS were not involved in CQAH-mediated apoptosis;
however, JNK inhibition by SP600125 significantly reduced
CQAH-induced downregulation of Bcl-XL and cell death. This finding
proved that JNK-dependent Bcl signaling participates in
CQAH-induced apoptosis. ROS are involved in various cellular
functions, including cell proliferation, differentiation, necrosis
and apoptosis. In addition, ROS can modify the mitochondrial
permeability, which induces the loss of the mitochondrial membrane
potential. As various anti-cancer drugs induce ROS-dependent
mitochondrial malfunction (52,53),
the present study used two effective anti-oxidants, NAC and DPI, to
identify whether ROS is involved in CQAH-induced cell death.
However, the results showed that pre-treatment with NAC and DPI did
not inhibit CQAH-mediated cell death or PARP and caspase-3
cleavage, which clearly demonstrated that neither hydrogen peroxide
generation nor superoxide generation is associated with CQAH
induced apoptosis and that ROS were not involved in JNK
activation.
In conclusion, the results of the present study
showed that substitution at position-4 of the phenyl ring attached
to azetidine affected the cytotoxicity of CQAH. As substitution at
other positions or moieties may lead to further optimization of the
activity of the compounds, the present study may serve as a basis
for structure-based drug design of quinoline derivatives for the
treatment of colon cancer. The present study identified that
CQAH-induced apoptosis was mediated via JNK activation and
caspases, while it was ROS-independent. Combined treatment with
chemotherapeutic drugs showed that CQAH enhances their therapeutic
efficacy against colon cancer. Therefore, CQAH and its derivatives
are a promising class of compounds which may be developed into
novel drugs for the treatment of colon cancer.
Acknowledgments
The present study was supported by the Natural
Science Foundation of Liaoning Province (grant no. 2014023028) and
the National Natural Science Foundation of China (grant no.
81273919).
References
|
1
|
Fearon ER and Vogelstein B: Genetic model
for colorectal cancer tumorigenesis. Cell. 61:759–767. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fearon ER and Jones PA: Progressing toward
a molecular description of colorectal cancer development. FASEB J.
6:2783–2790. 1992.PubMed/NCBI
|
|
3
|
Radtke F and Clevers H: Self-renewal and
cancer of the gut: two sides of a coin. Science. 307:1904–1909.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nelson H, Petrilli N, Carllin A, Coutoure
J, Fleshman J, Guillem J, Miedema B, Ota D and Sargent D: National
Cancer Institute Expert Panel: Guidelines 2000 for colon and rectal
cancer surgery. J Natl Cancer Inst. 93:583–596. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
O'Connell JB, Maggard MA and Ko CY: Colon
cancer survival rates with the new American joint committee on
cancer sixth edition tagging. J Natl Cancer Inst. 96:1420–1425.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Folprecht G, Grothey A, Alberts S, Raab HR
and Köhne CH: Neoadjuvant treatment of unresectable colorectal
liver metastases: Correlation between tumour response and resection
rates. Ann Oncol. 16:1311–1319. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Edwards MS, Chadda SD, Zhao Z, Barber BL
and Sykes DP: A systematic review of treatment guidelines for
metastatic colorectal cancer. Colorectal Dis. 14:e31–e47. 2012.
View Article : Google Scholar
|
|
8
|
Thun MJ, Namboodiri MM and Heath CW Jr:
Aspirin use and reduced risk of fatal colon cancer. N Engl J Med.
325:1593–1596. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Thun MJ, Namboodiri MM, Calle EE, Flanders
WD and Heath CW Jr: Aspirin use and risk of fatal cancer. Cancer
Res. 53:1322–1327. 1993.PubMed/NCBI
|
|
10
|
Marnett LJ: Aspirin and the potential role
of prostaglandins in colon cancer. Cancer Res. 52:5575–5589.
1992.PubMed/NCBI
|
|
11
|
Marnett LJ: Aspirin and related
nonsteroidal anti-inflammatory drugs as chemopreventive agents
against colon cancer. Prev Med. 24:103–106. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Giovannucci E, Rimm EB, Stampfer MJ,
Colditz GA, Ascherio A and Willett WC: Aspirin use and the risk for
colorectal cancer and adenoma in male health professionals. Ann
Intern Med. 121:241–246. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Giovannucci E, Egan KM, Hunter DJ,
Stampfer MJ, Colditz GA, Willett WC and Speizer FE: Aspirin and the
risk of colorectal cancer in women. N Engl J Med. 333:609–614.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Waddell WR and Loughry RW: Sulindac for
polyposis of the colon. J Surg Oncol. 24:83–87. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Waddell WR, Ganser GF, Cerise EJ and
Loughry RW: Sulindac for polyposis of the colon. Am J Surg.
157:175–179. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Giardiello FM, Hamilton SR, Krush AJ,
Piantadosi S, Hylind M, Celano P, Booker SV, Robinson CR and
Offerhaus GJ: Treatment of colonic and rectal adenomas with
sulindac in familial adenomatous polyposis. N Engl J Med.
328:1313–1316. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Giardiello FM, Offerhaus GJ and DuBois RN:
The role of nonsteroidal anti-inflammatory drugs in colorectal
cancer prevention. Eur J Cancer. 31A:1071–1076. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hare JI, Neijzen RW, Anantha M, Dos Santos
N, Harasym N, Webb MS, Allen TM, Bally MB and Waterhouse DN:
Treatment of colorectal cancer using a combination of liposomal
irinotecan (Irinophore C™) and 5-fluorouracil. PLoS One.
8:e623492013. View Article : Google Scholar
|
|
19
|
Allen WL, Coyle VM, Jithesh PV, Proutski
I, Stevenson L, Fenning C, Longley DB, Wilson RH, Gordon M, Lenz HJ
and Johnston PG: Clinical determinants of response to
irinotecan-based therapy derived from cell line models. Clin Cancer
Res. 14:6647–6655. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Troiani T, Serkova NJ, Gustafson DL,
Henthorn TK, Lockerbie O, Merz A, Long M, Morrow M, Ciardiello F
and Eckhardt SG: Investigation of two dosing schedules of
vandetanib, ZD6474, an inhibitor of vascular endothelial growth
factor receptor and epidermal growth factor receptor signaling, in
combination with irinotecan in a human colon cancer xenograft
model. Clin Cancer Res. 13:6450–6458. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pancreach E, Guérin E, Nicolet C,
Lelong-Rebel I, Voegeli AC, Oudet P, Larsen AK, Gaub MP and Guenot
D: Marked activity of irnotecan and rapamycin combination toward
colon cancer cells in vivo and in vitro is mediated through
cooperative modulation of the mammalian target of rapamycin/hypoxia
inducible factor-1apha axis. Clin Cancer Res. 15:1297–1307. 2009.
View Article : Google Scholar
|
|
22
|
Chen YL, Lin PC, Chen SP, Lin CC, Tsai NM,
Cheng YL, Chang WL, Lin SZ and Harn HJ: Activation of nonsteroidal
anti-inflammatory drug-activated gene-1 via extracellular
signal-regulated kinase 1/2 mitogen-activated protein kinase
revealed a isochaihulactone-triggered apoptotic pathway in human
lung cancer A549 cells. J Pharmacol Exp Ther. 323:746–756. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Egan TJ: Interactions of quinoline
antimalarials with hematin in solution. J Inorg Biochem.
100:916–926. 2006. View Article : Google Scholar
|
|
24
|
Abonia R, Insuasty D, Castillo J, Insuasty
B, Quiroga J, Nogueras M and Cobo J: Synthesis of novel
quinoline-2-one based chalcones of potential anti-tumor activity.
Eur J Med Chem. 57:29–40. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dürckheimer W, Blumbach J, Lattrell R and
Scheunemann KH: Recent developments in the field of β-Lactam
antibiotics. Angew Chem Int Ed Engl. 24:180–202. 1985. View Article : Google Scholar
|
|
26
|
Khalafallah AK, Selim MA, El-Hamd RMA,
Elmaghraby MA, Soleiman, et al: Novel synthesis of some new
fused/spiro heterocyclic compounds and their biological activity.
Indian J Chem. 34:1066–1070. 1995.
|
|
27
|
Parikh KA, Oza PS and Parikh AR: Synthesis
of some new 2-azetidinones as potential antitubercular agents.
Indian J Chem. 39B:7162000.
|
|
28
|
Patel NB and Patel JC: Synthesis and
antimicrobial activity of Schiff bases and 2-azetidinones derived
from quinazolin-4(3H)-one. Arabian J Chem. 4:403–411. 2011.
View Article : Google Scholar
|
|
29
|
Parmar SJ and Patel JI: Synthesis and
biological evaluation of some novel optically
active3-chloro-1-[4-({4-[(S)-(4-chlorophenyl)
(phenyl)methyl]-1-piperazinyl}acetyl)phenyl]-4-aryl-2-azetidin-onederivatives.
Der Pharma Chemica. 2:141–151. 2010.
|
|
30
|
Mathew B, Elizebeth MG, Mathew N and
Vijayabaskaran M: Synthesis, Characterisation of some 2-azetidinone
derivatives from 2-aminopyridine and evaluation of their
antimicrobial activity. Der Pharma Chemica. 6:238–242. 2012.
|
|
31
|
Vashi BS, Mehta DS and Shah VH: Synthesis
and biological activity of 4-thiazolidinones, 2-azetidinones,
4-imidazolinone derivatives having thymol moiety. Indian J Chem.
34B:802–808. 1995.
|
|
32
|
Polanski J, Niedbala H, Musiol R, Podeszwa
B, Tabak D, et al: 5-Hydroxy-8-nitro-6-quinaldic acid as a novel
molecular scaffold for HIV-1 integrase inhibitors. Lett Drugs Des
Disc. 3:175–178. 2006. View Article : Google Scholar
|
|
33
|
Polanski J, Niedbala H, Musiol R, Podeszwa
B, Tabak D, et al: Fragment based approach for the investigation of
HIV-1 integrase inhibition. Lett Drugs Des Disc. 4:99–105. 2007.
View Article : Google Scholar
|
|
34
|
Musiol R, Tabak D, Niedbala H, Podeszwa B,
Jampilek J, Kralova K, Dohnal J, Finster J, Mencel A and Polanski
J: Investigating biological activity spectrum for novel
quinolineanalogues 2: Hydroxyquinolinecarboxamides with
photosynthesis inhibiting activity. J Bioorg Med Chem.
16:4490–4499. 2008. View Article : Google Scholar
|
|
35
|
Jampilek J, Musiol R, Pesko M, Kralova K,
Vejsova M, Carroll J, Coffey A, Finster J, Tabak D, Niedbala H, et
al: Ring-substituted 4-Hydroxy-1H-quinolin-2-ones: Preparation and
biological activity. J Molecules. 14:1145–1159. 2009. View Article : Google Scholar
|
|
36
|
Patel R, Kumari P and Chikhalia K: Novel
s-Triazinyl piper-azines: Design, synthesis, characterization and
anti-microbial activity. Archives of Applied Science Research.
6:232–240. 2010.
|
|
37
|
Mistry B and Jauhari S: Synthesis and
characterization of some quinoline based azetidinones and
thiazolidinones as antimicrobial agents. Archives of Applied
Science Research. 6:332–343. 2010.
|
|
38
|
Parmar SJ and Patel IJ: Synthesis and
biological evaluation of some novel optically
active3-chloro-1-[4-({4-[(S)-(4-chlorophenyl)
(phenyl)methyl]-1-piperazinyl}acetyl)phenyl]-4-aryl-2-azetidin-onederivatives.
Der Pharma Chemica. 1:141–151. 2010.
|
|
39
|
Madhu G, Jayaveera KN, Ravindra Nath LK,
Kumar BS and Nagarjuna Reddy P: Synthesis and structure activity
relationship of new antibacterial active multi substituted
quinoline-azetidone mannich bases. Der Pharma Chemica. 3:1033–1040.
2012.
|
|
40
|
Johnson GL and Lapadat R:
Mitogen-activated protein kinase pathways mediated by ERK, JNK, and
p38 protein kinases. Science. 298:1911–1912. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ishihara Y, Matsunaga K, Iijima H,
Hasegawa G, Suzuki T, Sato A, Kobayashi T, Yang M and Hoffman RM:
The combination of 5-FU, leucovorin and CPT-11 (FOLFIRI) prolongs
survival through inhibition of metastasis in an orthotopic model of
colon cancer. Anticancer Res. 30:403–408. 2010.PubMed/NCBI
|
|
42
|
Zhang W and Liu HT: MAPK signal pathways
in the regulation of cell proliferation in mammalian cells. Cell
Res. 12:9–18. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Olsson H, Sjö P, Ersoy O, Kristoffersson
A, Larsson J and Nordén B: 4-Anilino-6-phenyl-quinoline inhibitors
of mitogen activated protein kinase-activated protein kinase 2
(MK2). Bioorg Med Chem Lett. 20:4738–4740. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang X, Mader MM, Toth JE, Yu X, Jin N,
Campbell RM, Smallwood JK, Christe ME, Chatterjee A, Goodson T Jr,
et al: Complete inhibition of anisomycin and UV radiation but not
cytokine induced JNK and p38 activation by an aryl-substituted
hydropyrrolopyrazole quinoline and mixed lineage kinase 7 small
interfering RNA. J Biol Chem. 280:19298–19305. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhu BK, Wang P, Zhang XD, Jiang CC, Chen
LH, Avery-Kiejda KA, Watts R and Hersey P: Activation of Jun
N-terminal kinase is a mediator of vincristine-induced apoptosis of
melanoma cells. Anticancer Drugs. 19:189–200. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Muscarella DE and Bloom SE: The
contribution of c-Jun N-terminal kinase activation and subsequent
Bcl-2 phosphorylation to apoptosis induction in human B-cells is
dependent on the mode of action of specific stresses. Toxicol Appl
Pharmacol. 228:93–104. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kelkel M, Cerella C, Mack F, Schneider T,
Jacob C, Schumacher M and Diederich M and Diederich M:
ROS-independent JNK activation and multisite phosphorylation of
Bcl-2 link diallyl tetra-sulfide-induced mitotic arrest to
apoptosis. Carcinogenesis. 33:2162–2171. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Leung KT, Li KK, Sun SS, Chan PK, Ooi VE
and Chiu LC: Activation of JNK pathway promotes phosphorylation and
degradation of BimEL-a novel mechanism of chemoresistance in T-cell
acute lymphoblastic leukemia. Carcinogenesis. 29:544–551. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Donovan N, Becker EB, Konishi Y and Bonni
A: JNK phosphorylation and activation of BAD couples the
stress-activated signaling pathway to the cell death machinery. J
Biol Chem. 277:40944–40949. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Han MH, Kim GY, Yoo YH and Choi YH:
Sanguinarine induces apoptosis in human colorectal cancer HCT-116
cells through ROS-mediated Egr-1 activation and mitochondrial
dysfunction. Toxicol Lett. 220:157–166. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhao WX, Tang SS, Jin X, et al:
Olaquindox-induced apoptosis is suppressed through p38 MAPK and
ROS-mediated JNK pathways in HepG2 cells. Cell Biol Toxicol.
29:229–238. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kuo YF, Su YZ, Tseng YH, Wang SY, Wang HM
and Chueh PJ: Flavokawain B, a novel chalcone from Alpinia pricei
Hayata with potent apoptotic activity: Involvement of ROS and
GADD153 upstream of mitochondria-dependent apoptosis in HCT116
cells. Free Radic Biol Med. 49:214–226. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Quan Z, Gu J, Dong P, Lu J, Wu X, Wu W,
Fei X, Li S, Wang Y, Wang J and Liu Y: Reactive oxygen
species-mediated endoplasmic reticulum stress and mitochondrial
dysfunction contribute to cirsimaritin-induced apoptosis in human
gallbladder carcinoma GBC-SD cells. Cancer Lett. 295:252–259. 2010.
View Article : Google Scholar : PubMed/NCBI
|