Introduction
Acetylation or deacetylation of histone proteins is
regulated by histone acetyltransferase (HAT) or histone deactylase
(HDAC), respectively. The equilibrium between HAT and HDAC acts as
a switch controlling the level of gene transcription, including
that of genes coding for inflammatory cytokines (1). HAT coordinates the recruitment and
activation of transcription factors by inducing conformational
changes in histones, allowing for access to gene promoters. HDAC
counteracts HAT activity by targeting of histones as well as
non-histone signal transduction proteins which have roles in
inflammation (2). Conditional
deletion of HDAC1 in T cells leads to enhanced airway inflammation
and increases in the synthesis of T-helper type 2 cell cytokine
production (3). The finding that
HDAC activity was depressed in synovial tissues from patients with
rheumatoid arthritis indicated that strategies restoring HDAC
function may have a therapeutic value in this disease (2).
Conversely, inhibition of HDAC with HDAC inhibitors
was demonstrated to limit the production of pro-inflammatory
cytokines, including tumor necrosis factor (TNF)-1α (4), and the expression of the sirtuin 1
gene is regulated by nuclear factor (NF)-κB, which is activated by
TNF-1α (5). Of note,
pharmacological inhibitors of HDAC activity have demonstrated
potent therapeutic effects in animal models of arthritis and other
chronic inflammatory diseases (6,7). A
recent study reported a markedly elevated HAT/HDAC ratio in
rheumatoid arthritis (RA) and ankylosing spondylitis (AS) during
anti-TNF-α therapy, while rituximab increased HAT as well as HDAC
(8). Previous studies have
reported an imbalance between HAT and HDAC in peripheral blood
mononuclear cells (PBMCs) or synovial tissues from patients with RA
and AS (9,10).
AS is a chronic inflammatory type of arthritis
affecting the axial as well as peripheral skeletons and soft
tissues. Changes in the expression of microRNA (miRNA) have been
demonstrated to be involved in the pathogenesis of various types of
arthritis, including RA and osteoarthritis (OA) (11,12).
A number of studies have shown that altered miRNA expression in
synovia, PBMCs or T cells from patients with RA or OA is linked
with innate immunity and inflammation (13–15).
It was recently demonstrated that miR-16, miR-221 and let-7i are
overexpressed in T cells from patients with AS, and mechanistic
studies showed that the increased let-7i expression facilitates the
T helper type 1, interferon (IFN)-γ-associated immune response in T
cells (16). Bioinformatics
analyses are widely used to identify potential targets of miR-130a
in endothelial progenitor cells (17), hepatitis C virus (18) and cardiomyocytes (19,20).
To date, the underlying mechanisms of miR-130a regulation in PBMCs
from patients with AS have largely remained elusive.
Advances in the development of effective therapies
for AS have been limited as the underlying mechanisms of AS causing
immune and inflammatory responses have not yet been elucidated.
Therefore, revealing the molecular mechanisms of AS is
indispensable for developing effective treatments. In the present
study, PBMCs were used investigate the pathogenesis of AS through
miR-130a via HDAC-associated pathways.
Materials and methods
Peripheral blood samples and cell
culture
Human peripheral blood samples were obtained with
written informed patient consent from the Department of
Orthopedics, The Thrid People's Hospital of Hefei (Hefei, China).
The present study was approved by the Ethics Committee of the
Department of Orthopedics, The Thrid People's Hospital of Hefei.
Peripheral blood samples from 20 AS patients and 20 normal healthy
control subjects were collected between February 2013 and December
2014.
The human PBMCs were separated from the peripheral
blood samples at the Cell Resource Center, Shanghai Institutes for
Biological Sciences (Shanghai, China), and maintained in RPMI-1640
(Invitrogen Life Technologies, Inc., Carlsbad, CA, USA)
supplemented with 10% fetal bovine serum (Invitrogen Life
Technologies) at 37°C in a humidified incubator (Thermo Fisher
Scientific, Waltham, MA, USA), in an atmosphere of 5%
CO2 and 95% air. The medium was replenished every
day.
Hierarchical cluster analysis
Microarray date were obtained from the Gene
Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/), using the GEO
accession number GSE25101. Observations with adjusted P-values
≥0.05 were removed, and thus excluded from further analysis. The
hierarchical cluster analysis was created using a method of
hierarchical clustering by GeneSpring GX, version 7.3 (Agilent
Technologies, Santa Clara, CA, USA).
RNA interference
The small interfering (si) RNA for human HDAC3 and
scramble siRNA were obtained from Dharmacon (Lafayette, CO, USA).
The following primers were used:HDAC3 forward,
5′-CACUCUGAGUGGGACAAGCUCUUCA-3′ and reverse,
5′-UGAAGAGCUUGUCCCACUCAGAGUG-3′; miRNA-130a forward,
5′-GCUAUCAGUCCACUGUGCU UGUGGU-3′ and reverse,
5′-ACCACAAGCACAGUGGACU GAUAGC-3′; scramble forward,
5′-CACGAGUGGGUAACACUCGUCUUCA-3′ and reverse,
5′-UGAAGACGAGUGUUACCCACUCGUG-3′. The siRNA oligonucleotides (at a
final concentration of 100 nM) were transfected into human PBMCs
using Lipofectamine 2000 (Invitrogen Life Technologies) according
to the manufacturer's instructions. Trichostatin A (TSA) and
dimethyl sulfoxide (DMSO) was obtained from Sigma-Aldrich (cat nos.
V900931 and D2650; Sigma-Aldrich, St. Louis, MO, USA). The HDAC3
inhibitor TSA and negative control DMSO were used at a
concentration of 25 µM for 5 h when the cells had reached
60–80% confluence.
miRNA mimic and transfection
The human miR-130a duplex mimic (miR-130a mimic) and
negative control oligonucleotide duplex mimic (miR-NC) were
designed and synthesized by Guangzohu RiboBio Co., Ltd. (Guangzhou,
China). miR-130a mimic sequence: 5′-CAGUGCAAUGUUAAAAGGG-3′; and
miR-NC sequence: 5′-UUCUCCGAACGUGUCACGUTT-3′ Once cells reached
30–50% confluence they were transfected with 1 nM miRNAs using
Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA), according to
the manufacturer's protocol. Total RNA was extracted 24 h
post-transfection, and total cell protein was extracted 48 or 72 h
post-transfection.
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR)
RNA extraction from PBMCs was performed with TRIzol
(Invitrogen Life Technologies) according to the manufacturer's
instructions. Synthesis of cDNA was performed by reverse
transcription reaction with 2 µg total RNA using moloney
murine leukemia virus reverse transcriptase (Promega, Madison, WI,
USA) with oligo dT (15) primers
(Fermentas; Thermo Fisher Scientific) according to the
manufacturer's instructions. The first-strand cDNAs served as the
template for the regular PCR performed using
TransScript® II Green Two-Step qRT-PCR SuperMix
(Transgen Biotech Co., Ltd., Beijing, China), which included
EasyTaq® DNA polymerase, dNTPs and buffer. The cycling
conditions were as follows: 2 min of polymerase activation at 95°C
followed by 40 cycles at 95°C for 15 sec, 55°C for 60 sec and 72°C
for 20 sec. PCR was performed using the following primers: HDAC3
forward, 5′-TTGCGATTCTGTTTTGTGCT-3′ and reverse,
5′-GTGGGGTCCTCAGTGGG-3′; miRNA-130a forward,
5′-TTGCGATTCTGTTTTGTGCT-3′ and reverse, 5′-GTGGGGTCCTCAGTGGG-3′;
β-actin forward, 5′-ACAGGGGAGGTGATAGCATT-3′ and reverse,
5′-GACCAAAAGCCTTCATACATCTC-3′. All primers were purchased from
Sangon Biotech Co., Ltd. (Shanghai, China). β-actin as an internal
control was used to normalize the data to determine the relative
expression of the target genes. RT-qPCR was carried out on a
LightCycler 480 (Roche Diagnostics GmbH, Penzberg, Germany). After
completion of the reaction, the amplification curve and melting
curve were analyzed. Gene expression values were determined using
the 2−ΔΔCt method.
Western blot analysis
The PBMCs were homogenized and extracted with NP-40
buffer (Beyotime Institute of Biotechnology, Haimen, China),
followed by 5–10 min boiling and centrifugation (10,500 × g, 15
min, 4°C) to obtain the supernatant. Samples containing 50
µg protein were separated by 10% SDS-PAGE and transferred
onto a polyvinylidene difluoride Transfer Membrane (Millipore,
Billerica, MA, USA). After saturation with 5% (w/v) non-fat dry
milk in Tris-buffered saline and 0.1% (w/v) Tween 20 (TBST), the
membranes were incubated with the rabbit anti-human polyclonal
HDAC3 (cat. no. ab16047) and mouse anti-human monoclonal TNF-1α
(cat. no. ab10204) antibodies (Abcam, Cambridge, MA, USA) at
dilutions of 1:500–1:2,000 at 4°C overnight. After three washes
with TBST, membranes were incubated with secondary immunoglobulins
(Igs) conjugated to horseradish peroxidase, including goat
anti-mouse IgG (cat. no. ab6789) and goat anti-rabbit IgG (cat. no.
ab6721) (Abcam) at a dilution of 1:10,000 and 1:20,000. After 1 h
of incubation at 37°C, membranes were washed three times with TBST.
Blots were visualized using the Odyssey Infrared Imaging System
(LI-COR Biosciences). Signals were densitometrically assessed
(Odyssey Application Software version 3.0; LI-COR Biosciences) and
normalized to the β-actin signals to correct for unequal loading
using the mouse monoclonal anti-β-actin antibody (1:10,000
dilution; cat. no. ab6276; Abcam).
Chromatin immunoprecipitation (ChIP)
assay
PBMCs were cross-linked with 0.5% formaldehyde
(Beyotime Institute of Biotechnology) for 10 min at room
temperature. Cross-linking was stopped by adding 125 mM glycine
(Beyotime Institute of Biotechnology). Cells were solubilized in a
buffer containing 10 mM Tris-HCl (pH 8.0), 1% Triton X-100, 1%
sodium deoxy-cholate, 1 mM phenylmethanesulfonylfluoride and
protease inhibitor cocktail (all Beyotime Institute of
Biotechnology) for 10 min at 4°C. Pellets obtained by
centrifugation at 10,006 × g for 5 min were suspended in
radioimmunoprecipitation assay buffer (Beyotime Institute of
Biotechnology) and sonicated using a BioruptorSonicator (Diagenode,
Seraing, Belgium) to shear chromatin into 500-bp fragments. The
immunoprecipitated protein-DNA complexes were collected using
protein A agarose beads (Upstate Biotechnology, Inc., Lake Placid,
NY, USA). Sonicated chromatin was subjected to immunoprecipitation
using ChIP-grade agarose beads with protein G (Cell Signaling
Technology, Inc., Beverly, MA, USA), blocked with 1% bovine serum
albumin (BSA; Beyotime Institute of Biotechnology) and 1% salmon
sperm DNA (Beyotime Institute of Biotechnology). Proteinase K (1
mg/ml, Sigma-Aldrich) digestion and DNA purification (Wizard Plus
Minipreps DNA Purification system; Promega) were performed after
blocking with BSA and salmon sperm DNA. The resultant product was
then subjected to PCR amplification.
Statistical analysis
Values are expressed as the mean ± standard error of
mean for each group. All statistical analyses were performed by
using GraphPad Prism version 4.0 (GraphPad Inc., La Jolla, CA,
USA). One-way analysis of variance or Student's t-tests were
applied, as well as regression analysis to analyze data.
Differences with a P-value of <0.05 were considered
statistically significant.
Results
HDAC3 expression is increased and
miRNA-130a expression is decreased in PBMCs from AS patients
A recent study has shown that in PBMC nuclear
extracts from AS patients, the levels of HAT were decreased, while
HDAC tended to be increased compared with that in healthy control
subjects (8). Consistent with this
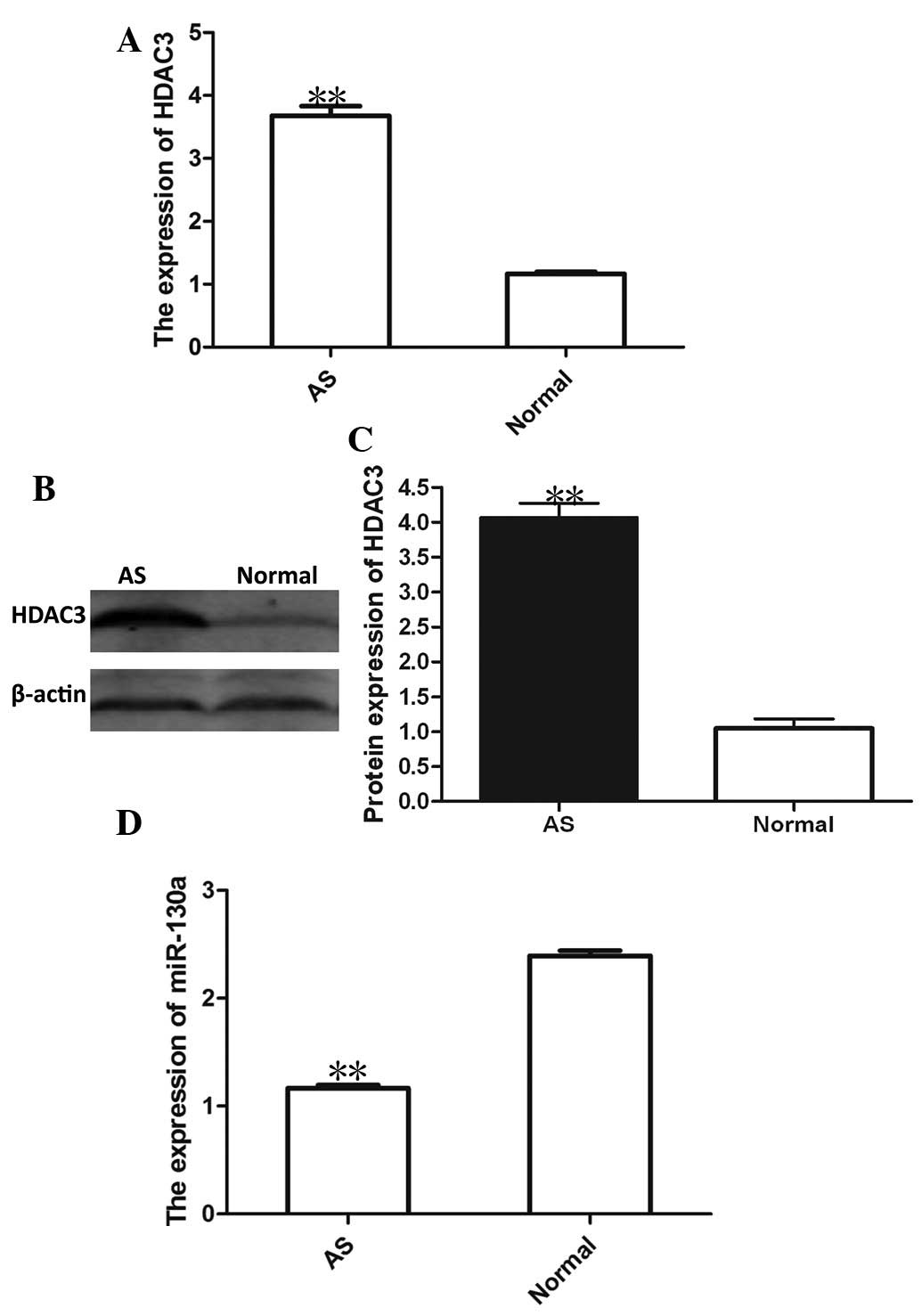
previous study, the results of the present study showed a
>3-fold increase in HDAC3 mRNA and protein expression in PBMCs
from patients with AS compared with that in healthy control
subjects (Fig. 1A–C). The present
study then investigated the possible link between miRNA expression
and AS. A hierarchical cluster analysis of differentially expressed
miRNAs in the PBMCs from AS patients was performed. The results
showed that the down-regulation of miRNA-130a was associated with
AS, the expression of which was lowest among all miRNAs. As shown
in Fig. 1D, miRNA-130a expression
was markedly decreased in PBMCs from AS patients compared with that
in PBMCs from normal controls.
miRNA-130a is regulated by HDAC3
The deregulation of miRNAs has been frequently
identified in immunopathogenesis and has been linked to AS
(16,21). The expression of
miRNA-15a/miRNA-16-1 is repressed through recruitment of HDAC3 in
mantle cells and other non-Hodgkin B-cell lymphomas (21). Based on these data, the present
study investigated whether expression of miRNA-130a in PBMCs from
AS patients was affected by HDAC3 knockdown. Increased miRNA-130a
expression was observed in the HDAC3 siRNA group and in the HDAC3
inhibitor [trichostatin A (TSA)] group (Fig. 2A). Furthermore, a ChIP assay was
performed using HDAC3 antibody to detect binding at the miRNA-130a
promoter. This approach revealed that HDAC3 was able to bind to
miRNA-130a promoters in PBMCs and the percentage input was
significantly increased for AS patients compared with that for
normal controls (Fig. 2B). These
findings suggested that HDAC3-mediated histone hypoacetylation
contributed to the regulation of the expression of miRNA-130a.
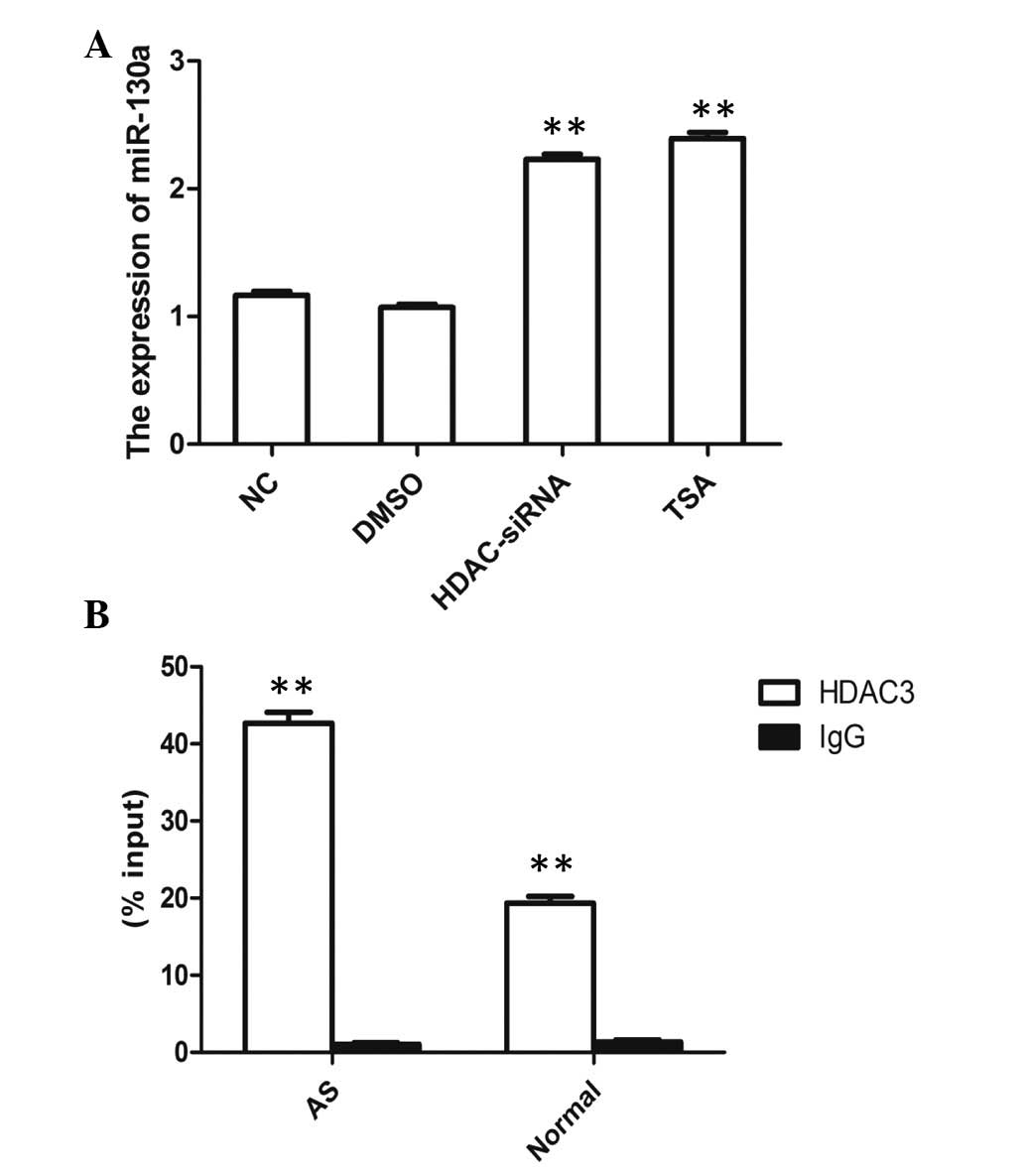 | Figure 2Regulation of miRNA-130a by HDAC3. (A)
Peripheral blood mononuclear cells were treated with vehicle
solvent, DMSO, HDAC-siRNA or TSA, and the expression of HDAC3 was
assessed by reverse transcription quantitative polymerase chain
reaction. **P<0.01 vs. NC. (B) A ChIp assay was
performed using HDAC3 antibody to detect binding at the miRNA-130a
promoter region. Percentage input was calculated using the formula
2(Ct[1% of input] − Ct[ChIP]). Values are expressed as
the mean ± standard error of the mean; n=3 per group.
**P<0.01 vs. IgG. DMSO, dimethyl sulfoxide; siRNA,
small interfering RNA; TSA, trichostatin A; ChIP, chromatin
immunoprecipitation; Ct, cycle threshold; AS, ankylosing
spondylitis; miRNA, microRNA; IgG, immunoglobulin G; NC, negative
control. |
TNF-1α is regulated by HDAC3 via
miRNA-130a
miR-130a overexpression led to a decrease, whereas
miR-130a inhibition caused an increase of the mRNA and protein
expression of TNF-1α in PBMCs (Fig. 3A
and B). Furthermore, HDAC3 knockdown or HDAC3 inhibition caused
an increased in the expression of miR-130a and a decrease of the
mRNA and protein expression of TNF-1α in PBMCs (Fig. 3C and D). These observations
suggested that HDAC3 has an important role in the underlying
mechanism of AS by regulating miR-130a expression via its target
TNF-1α.
 | Figure 3Regulation of TNF-1α by HDAC3 via
miRNA-130a. The (A) mRNA and (B) protein expression of TNF-1α in
PBMCs with miRNA-130a knockdown or overexpression were assessed by
RT-qPCR and western blot analysis, respectively. (C) The mRNA
expression of TNF-1α and miRNA-130a in PBMCs with HDAC3 knockdown
or inhibition with TSA was assessed by RT-qPCR. (D) The protein
expression of TNF-1α in PBMCs with HDAC3 knockdown or inhibition by
TSA was assessed by western blot analysis. Blots are representative
of three experiments. Values are expressed as the mean ± standard
error of the mean; n=3 per group. **P<0.01 vs.
miRNA-130a of the NC group; ##P<0.01 vs. TNF-1α of
the NC group. TNF, tumor necrosis factor; HDAC3, histone deactylase
3; miRNA, microRNA; PBMC, peripheral blood mononuclear cell;
RT-qPCR, reverse transcription quantitative polymerase chain
reaction; DMSO, dimethyl sulfoxide; TSA, trichostatin A; siRNA,
small interfering RNA; NC, negative control. |
Discussion
Among the numerous HDACs, HDAC3 is widely expressed
and conserved in a wide range of species (22). HDAC3 regulates the c-Jun N-terminal
kinase pathway (23), NF-κB
activity (24), mitogen-activated
protein kinase activation (25)
and nuclear translocation of regulator of calcineurin 1 (26). The present study reported several
important observations regarding the involvement of HDAC3 in the
molecular mechanisms of AS. First, increased mRNA and protein
levels of HDAC3 and decreased levels of miRNA-130a were observed in
PBMCs from AS patients. Furthermore, HDAC3 knockdown or HDAC3
inhibition promoted the expression of miRNA-130a and HDAC3 could be
recruited to the promoter region of the gene encoding for
miRNA-130a in AS patients. From these results, it can be deduced
that HDAC3 is involved in the regulation of a distinct underlying
molecular mechanism of AS by forming a negative feedback loop with
miR-130a. In addition, miR-130a overexpression led to a decrease,
whereas miR-130a inhibition caused an increase in TNF-1α expression
in PBMCs. Furthermore, HDAC3 knockdown or HDAC3 inhibition caused
an increase in the expression of miR-130a and a decrease in the
expression of TNF-1α in PBMCs. All of these observations suggested
that HDAC3 has an important role in the underlying molecular
mechanism of AS by regulating miR-130a expression via its target
TNF-1α.
A recent study demonstrated that miRNA-130a is
required for normal immune function. miRNA-130a was demonstrated to
markedly inhibit hepatitis virus C (HCV) replication in the HCV
replicon as well as in J6-/JFH1-infected cells. Overexpression of
miR-130a led to increases in the expression of type I IFN
(IFN-α/IFN-β), interferon-induced 17 kDa protein,
ubiquitin-specific peptidase 18 and MX dynamin-like guanosine
triphosphatase A, which are involved in the innate immune response;
furthermore, miR-130A overexpression led to decreases in the
expression of miR-122, a well-defined miRNA promoting HCV
production (18). miRNAs are
potentially differentially expressed in the T cells of AS patients,
which may alter the expression of the downstream target molecules
that may contribute to the pathogenesis of AS (16). In the present study, miRNA-130a
inhibition increased the expression of TNF-1α, while miRNA-130a
knockdown decreased the expression of TNF-1α. These results
indicated that TNF-1α may be a candidate target of miRNA-130a, as
it appeared to have an important regulatory role in the underlying
mechanisms of AS.
AS therapy has been revolutionized by the increasing
knowledge of the pathogenetic mechanisms of the disease, involving
dysfunction and excessive secretion of multiple pro-inflammatory
molecules, in particular TNF-1α (16,27).
Among five biological agents currently used in AS therapies,
anti-TNF-1α monoclonal antibody adalimumab, TNF receptor fusion
protein etanercept and anti-TNF-1α monoclonal antibody infliximab
have been largely demonstrated to be effective at reducing AS
activity and controlling joint damage and various aspects of the
diseases as well as to be reasonably safe (27–29).
Clinical studies have indicated that AS patients respond to the
above treatments. Upon TNF-1α inhibition, HAT activity increases
considerably in patients with AS (by 198%), leading to a marked
increase of the HAT/HDAC ratio in AS patients during anti-TNF-1α
therapy. The HDAC inhibitor TSA induces a decline in HDAC (by
43.6%) (8). In the present study,
the HDAC3-inhibitory function of TSA appeared to directly regulate
the expression of miRNA-130a as well as TNF-1α. Therefore, the
present study identified the HDAC3/miRNA-130a/TNF-1α axis as a
novel regulator of the pathogenetic mechanisms of AS.
In conclusion, the present study identified that
HDAC3 had an important role in the underlying molecular mechanism
of AS by regulating miR-130a expression via its target TNF-1α.
These results suggested that HDAC3, as an inflammatory mediator
which was shown to be elevated in PBMCs of patients with AS, may,
at least in part, contribute to the pathogenetic mechanism of AS.
Furthermore, the present study indicated that HDAC3 may represent a
therapeutic target for the treatment of AS.
References
|
1
|
Kim N, Sun HY, Youn MY and Yoo JY:
IL-1β-specific recruitment of GCN5 histone acetyltransferase
induces the release of PAF1 from chromatin for the de-repression of
inflammatory response genes. Nucleic Acids Res. 41:4495–4506. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Grabiec AM, Tak PP and Reedquist KA:
Targeting histone deacetylase activity in rheumatoid arthritis and
asthma as prototypes of inflammatory disease: Should we keep our
HATs on? Arthritis Res Ther. 10:2262008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Grausenburger R, Bilic I, Boucheron N,
Zupkovitz G, El-Housseiny L, Tschismarov R, Zhang Y, Rembold M,
Gaisberger M, Hartl A, et al: Conditional deletion of histone
deacetylase 1 in T cells leads to enhanced airway inflammation and
increased Th2 cytokine production. J Immunol. 185:3489–3497. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Roger T, Lugrin J, Le Roy D, Goy G,
Mombelli M, Koessler T, Ding XC, Chanson AL, Reymond MK, Miconnet
I, et al: Histone deacetylase inhibitors impair innate immune
responses to Toll-like receptor agonists and to infection. Blood.
117:1205–1217. 2011. View Article : Google Scholar
|
|
5
|
Katto J, Engel N, Abbas W, Herbein G and
Mahlknecht U: Transcription factor NFkB regulates the expression of
the histone deacetylase SIRT1. Clin Epigenetics. 5:112013.
View Article : Google Scholar
|
|
6
|
Blanchard F and Chipoy C: Histone
deacetylase inhibitors: New drugs for the treatment of inflammatory
diseases? Drug Discov Today. 10:197–204. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Choi JH, Oh SW, Kang MS, Kwon HJ, Oh GT
and Kim DY: Trichostatin A attenuates airway inflammation in mouse
asthma model. Clin Exp Allergy. 35:89–96. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Toussirot E, Wendling D and Herbein G:
CIC-1431: Biological treatments given in patients with rheumatoid
arthritis or ankylosing spondylitis modify HAT/HDAC (histone
acetyl-transferase/histone deacetylase) balance. Joint Bone Spine.
81:544–545. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gillespie J, Savic S, Wong C, Hempshall A,
Inman M, Emery P, Grigg R and McDermott MF: Histone deacetylases
are dysregulated in rheumatoid arthritis and a novel histone
deacetylase 3-selective inhibitor reduces interleukin-6 production
by peripheral blood mononuclear cells from rheumatoid arthritis
patients. Arthritis Rheum. 64:418–422. 2012. View Article : Google Scholar
|
|
10
|
Strietholt S, Maurer B, Peters MA, Pap T
and Gay S: Epigenetic modifications in rheumatoid arthritis.
Arthritis Res Ther. 10:2192008. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang CR, Shih KS, Liou JP, Wu YW, Hsieh
IN, Lee HY, Lin TC and Wang JH: Denbinobin upregulates miR-146a
expression and attenuates IL-1β-induced upregulation of ICAM-1 and
VCAM-1 expressions in osteoarthritis fibroblast-like synoviocytes.
J Mol Med (Berl). 2014.Epub ahead of print. View Article : Google Scholar
|
|
12
|
Lu MC, Yu CL, Chen HC, Yu HC, Huang HB and
Lai NS: Increased miR-223 expression in T cells from patients with
rheumatoid arthritis leads to decreased insulin-like growth
factor-1-mediated interleukin-10 production. Clin Exp Immunol.
177:641–651. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fulci V, Scappucci G, Sebastiani GD,
Giannitti C, Franceschini D, Meloni F, Colombo T, Citarella F,
Barnaba V, Minisola G, et al: miR-223 is overexpressed in
T-lymphocytes of patients affected by rheumatoid arthritis. Hum
Immunol. 71:206–211. 2010. View Article : Google Scholar
|
|
14
|
Pauley KM, Satoh M, Chan AL, Bubb MR,
Reeves WH and Chan EK: Upregulated miR-146a expression in
peripheral blood mononuclear cells from rheumatoid arthritis
patients. Arthritis Res Ther. 10:R1012008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhu S, Pan W, Song X, Liu Y, Shao X, Tang
Y, Liang D, He D, Wang H, Liu W, et al: The microRNA miR-23b
suppresses IL-17-associated autoimmune inflammation by targeting
TAB2, TAB3 and IKK-α. Nat Med. 18:1077–1086. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lai NS, Yu HC, Chen HC, Yu CL, Huang HB
and Lu MC: Aberrant expression of microRNAs in T cells from
patients with ankylosing spondylitis contributes to the
immunopathogenesis. Clin Exp Immunol. 173:47–57. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Meng S, Cao J, Zhang X, Fan Y, Fang L,
Wang C, Lv Z, Fu D and Li Y: Downregulation of microRNA-130a
contributes to endothelial progenitor cell dysfunction in diabetic
patients via its target Runx3. PloS One. 8:e686112013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li S, Duan X, Li Y, Liu B, McGilvray I and
Chen L: MicroRNA-130a inhibits HCV replication by restoring the
innate immune response. J Viral Hepat. 21:121–128. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Osbourne A, Calway T, Broman M, McSharry
S, Earley J and Kim GH: Downregulation of connexin43 by
microRNA-130a in cardiomyocytes results in cardiac arrhythmias. J
Mol Cell Cardiol. 74:53–63. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim GH, Samant SA, Earley JU and Svensson
EC: Translational control of FOG-2 expression in cardiomyocytes by
microRNA-130a. PloS One. 4:e61612009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang X, Chen X, Lin J, Lwin T, Wright G,
Moscinski LC, Dalton WS, Seto E, Wright K, Sotomayor E and Tao J:
Myc represses miR-15a/miR-16-1 expression through recruitment of
HDAC3 in mantle cell and other non-Hodgkin B-cell lymphomas.
Oncogene. 31:3002–3008. 2012. View Article : Google Scholar
|
|
22
|
Kim Y, Kim H, Park H, Park D, Lee H, Lee
YS, Choe J, Kim YM and Jeoung D: miR-326-histone deacetylase-3
feedback loop regulates the invasion and tumorigenic and angiogenic
response to anti-cancer drugs. J Biol Chem. 289:28019–28039. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang J, Kalkum M, Chait BT and Roeder RG:
The N-CoR-HDAC3 nuclear receptor corepressor complex inhibits the
JNK pathway through the integral subunit GPS2. Mol Cell. 9:611–623.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bendinelli P, Matteucci E, Maroni P and
Desiderio MA: NF-kappaB activation, dependent on
acetylation/deacetylation, contributes to HIF-1 activity and
migration of bone metastatic breast carcinoma cells. Mol Cancer
Res. 7:1328–1341. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mahlknecht U, Will J, Varin A, Hoelzer D
and Herbein G: Histone deacetylase 3, a class I histone
deacetylase, suppresses MAPK11-mediated activating transcription
factor-2 activation and represses TNF gene expression. J Immunol.
173:3979–3990. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Han KA, Kang HS, Lee JW, Yoo L, Im E, Hong
A, Lee YJ, Shin WH and Chung KC: Histone deacetylase 3 Promotes
RCAN1 Stability and nuclear translocation. PloS One. 9:e1054162014.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fabbroni M, Cantarini L, Caso F, Costa L,
Pagano VA, Frediani B, Manganelli S and Galeazzi M: Drug retention
rates and treatment discontinuation among anti-TNF-α agents in
psoriatic arthritis and ankylosing spondylitis in clinical
practice. 2014:8629692014.
|
|
28
|
Fenix-Caballero S, Alegre-del Rey EJ,
Castaño-Lara R, Puigventós-Latorre F, Borrero-Rubio JM and
López-Vallejo JF: Direct and indirect comparison of the efficacy
and safety of adalimumab, etanercept, infliximab and golimumab in
psoriatic arthritis. J Clin Pharm Ther. 38:286–293. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Costa L, Caso F, Atteno M, Del Puente A,
Darda MA, Caso P, Ortolan A, Fiocco U, Ramonda R, Punzi L and
Scarpa R: Impact of 24-month treatment with etanercept, adalimumab,
or methotrexate on metabolic syndrome components in a cohort of 210
psoriatic arthritis patients. Clin Rheumatol. 33:833–839. 2014.
|